Паренхиматозные дистрофии
Паренхиматозные дистрофии - проявления нарушений обмена в высокоспециализированных в функциональном отношении клетках. Поэтому при паренхиматозных дистрофиях преобладают нарушения клеточных механизмов трофики. Различные виды паренхиматозных дистрофий отражают недостаточность определенного физиологического (ферментативного) механизма, служащего выполнению специализированной функции клеткой (гепатоцит, нефроцит, кардиомиоцит и т.д.). В связи с этим в разных органах (печень, почки, сердце и т.д.) при развитии одного и того же вида дистрофии участвуют различные пато- и морфогенетические механизмы. Из этого следует, что переход одного вида паренхиматозной дистрофии в другой вид исключается, возможно лишь сочетание разных видов этой дистрофии.
В зависимости от нарушений того или иного вида обмена паренхиматозные дистрофии делят на белковые (диспротеинозы), жировые (липидозы) и углеводные.
Паренхиматозные белковые дистрофии (диспротеинозы)
Большая часть белков цитоплазмы (простых и сложных) находится в соединении с липидами, образуя липопротеидные комплексы. Эти комплексы составляют основу мембран митохондрий, эндоплазматической сети, пластинчатого комплекса и других структур. Помимо связанных белков, в цитоплазме содержатся и свободные. Многие из последних обладают функцией ферментов.
Сущность паренхиматозных диспротеинозов состоит в изменении физико-химических и морфологических свойств белков клетки: они подвергаются денатурации и коагуляции или, наоборот, колликвации, что ведет к гидратации цитоплазмы; в тех случаях, когда нарушаются связи белков с липидами, возникает деструкция мембранных структур клетки. В исходе этих нарушений может развиться коагуляционный (сухой) иликолликвационный (влажный) некроз (схема I).
К паренхиматозным диспротеинозам относят гиалиново-капельную, гидропическую и роговую дистрофии.
К паренхиматозным белковым дистрофиям со времен Р. Вирхова причисляли и многие патологи продолжают причислять так называемую зернистую дистрофию, при которой в клетках паренхиматозных органов появляются белковые зерна. Сами органы увеличиваются в размерах, становятся дряблыми и тусклыми на разрезе, что послужило причиной называть также зернистую дистрофию тусклым (мутным) набуханием.Однако электронно-микроскопическое и гистоферменто-
Схема I.Морфогенез паренхиматозных диспротеинозов 59
химическое изучение «зернистой дистрофии» показало, что в ее основе лежит не накопление белка в цитоплазме, а гиперплазия ультраструктур клеток паренхиматозных органов как выражение функционального напряжения этих органов в ответ на различные воздействия; гиперплазированные ультраструктуры клетки выявляются при светооптическом исследовании как белковые гранулы.
Гиалиново-капельная дистрофия
При гиалиново-капельной дистрофии в цитоплазме появляются крупные гиалиноподобные белковые капли, сливающиеся между собой и заполняющие тело клетки; при этом происходит деструкция ультраструктурных элементов клетки. В ряде случаев гиалиново-капельная дистрофия завершается фокальным коагуляционным некрозом клетки.
Этот вид диспротеиноза часто встречается в почках, редко - в печени и совсем редко - в миокарде.
В почкахпри микроскопическом исследовании накопление гиалиновых капель находят в нефроцитах. При этом наблюдается деструкция митохондрий, эндоплазматической сети, щеточной каемки (рис. 27). В основе гиалиново-капельной дистрофии нефроцитов лежит недостаточность вакуолярно-лизосомального аппарата эпителия проксимальных канальцев, в норме реабсорбирующего белки. Поэтому этот вид дистрофии нефроцитов очень часто встречается при нефротическом синдроме. Этот синдром является одним из проявлений многих заболеваний почек, при которых первично поражается гломерулярный фильтр (гломерулонефрит, амилоидоз почек, парапротеинемическая нефропатия и др.).
Внешний вид почек при этой дистрофии не имеет каких-либо характерных черт, он определяется прежде всего особенностями основного заболевания (гломерулонефрит, амилоидоз).
В печенипри микроскопическом исследовании в гепатоцитах находят гиалиноподобные тельца (тельца Мэллори), которые состоят из фибрилл 60
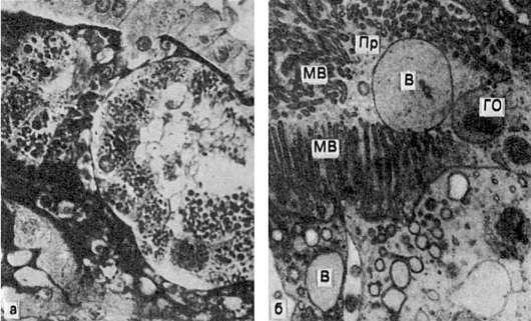
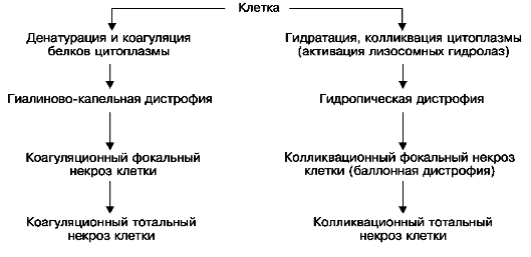
Рис. 27.Гиалиново-капельная дистрофия эпителия почечных канальцев:
а - в цитоплазме эпителия крупные белковые капли (микроскопическая картина); б - в цитоплазме клетки много белковых (гиалиновых) образований (ГО) овальной формы и вакуолей (В); отмечаются десквамация микроворсинок (МВ) щеточной каемки и выход в просвет (Пр) канальца вакуолей и белковых образований. Электронограмма. х18 000
особого белка - алкогольного гиалина (см. рис. 22). Образование этого белка и телец Мэллори служит проявлением извращенной белковосинтетической функции гепатоцита, что встречается постоянно при алкогольном гепатите и сравнительно редко при первичном билиарном и индийском детском циррозах, гепатоцеребральной дистрофии (болезни Вильсона-Коновалова).
Внешний вид печени различен; изменения характерны для тех ее заболеваний, при которых встречается гиалиново-капельная дистрофия.
Исход гиалиново-капельной дистрофии неблагоприятен: она завершается необратимым процессом, ведущим к некрозу клетки.
Функциональное значение этой дистрофии очень велико. С гиалиновокапельной дистрофией эпителия почечных канальцев связаны появление в моче белка (протеинурия) и цилиндров (цилиндрурия), потеря белков плазмы (гипопротеинемия), нарушение ее электролитного баланса. Гиалиново-капельная дистрофия гепатоцитов нередко является морфологической основой нарушений многих функций печени.
Гидропическая дистрофия
Гидропическая, или водяночная, дистрофия характеризуется появлением в клетке вакуолей, наполненных цитоплазматической жидкостью. Она наблюдается чаще в эпителии кожи и почечных канальцев, в гепатоцитах, мышечных и нервных клетках, а также в клетках коры надпочечников.
Микроскопическая картина:паренхиматозные клетки увеличены в объеме, цитоплазма их заполнена вакуолями, содержащими прозрачную жидкость. Ядро смещается на периферию, иногда вакуолизируется или сморщивается. Прогрессирование этих изменений приводит к распаду ультраструктур клетки и переполнению клетки водой. Клетка превращается в заполненные жидкостью баллоны или в огромную вакуоль, в которой плавает пузырьковидное ядро. Такие изменения клетки, которые по существу являются выражениемфокального колликвационного некроза называют баллонной дистрофией.
Внешний видорганов и тканей мало изменяется при гидропическои дистрофии, она обнаруживается обычно под микроскопом.
Механизм развитиягидропическои дистрофии сложен и отражает нарушения водно-электролитного и белкового обмена, ведущие к изменению коллоидно-осмотического давления в клетке. Большую роль играет нарушение проницаемости мембран клетки, сопровождающееся их распадом. Это ведет к закислению цитоплазмы, активации гидролитических ферментов лизосом, которые разрывают внутримолекулярные связи с присоединением воды.
Причиныразвития гидропической дистрофии в разных органах неоднозначны. В почках - это повреждение гломерулярного фильтра (гломерулонефрит, амилоидоз, сахарный диабет), что ведет к гиперфильтрации и недостаточности ферментной системы базального лабиринта нефроцитов, в норме обеспечивающей реабсорбцию воды; поэтому гидропическая дистрофия нефроцитов так характерна для нефротического синдрома. В печени гидропическая дистрофия возникает при вирусном и токсическом гепатитах (рис. 28) и нередко является причиной печеночной недостаточности. Причиной гидропическои дистрофии эпидермисаможет быть инфекция (оспа), отек кожи различного механизма. Вакуолизация цитоплазмы может быть проявлением физиологической деятельности клетки, что отмечается, например, в ганглиозных клетках центральной и периферической нервной системы.
Исходгидропической дистрофии, как правило, неблагоприятный; она завершается фокальным или тотальным некрозом клетки. Поэтому функция органов и тканей при гидропической дистрофии резко страдает.
Роговая дистрофия
Роговая дистрофия, или патологическое ороговение, характеризуется избыточным образованием рогового вещества в ороговевающем эпителии (гиперкератоз, ихтиоз) или образованием рогового вещества там, где в норме его не бывает (патологическое ороговение на слизистых оболочках, или лейкоплакия; образование «раковых жемчужин» в плоскоклеточном раке). Процесс может быть местным или распространенным.

Рис. 28.Гидропическая дистрофия печени (биопсия):
а - микроскопическая картина; вакуолизация гепатоцитов; б - электронограмма: расширение канальцев эндоплазматической сети и образование вакуолей (В), заполненных хлопьевидным содержимым. Мембраны, ограничивающие вакуоли, почти полностью лишены рибосом. Вакуоли сдавливают расположенные между ними митохондрии (М), часть которых подвергается деструкции; Я - ядро гепатоцита. х18 000
Причиныроговой дистрофии разнообразны: нарушение развития кожи, хроническое воспаление, вирусные инфекции, авитаминозы и др.
Исходможет быть двояким: устранение вызывающей причины в начале процесса может привести к восстановлению ткани, однако в далеко зашедших случаях наступает гибель клеток.
Значениероговой дистрофии определяется ее степенью, распространенностью и длительностью. Длительно существующее патологическое ороговение слизистой оболочки (лейкоплакия) может явиться источником развития раковой опухоли. Врожденный ихтиоз резкой степени, как правило, несовместим с жизнью.
К группе паренхиматозных диспротеинозов примыкает ряд дистрофий, в основе которых лежат нарушения внутриклеточного метаболизма ряда аминокислот в результате наследственной недостаточности метаболизирующих их ферментов, т.е. в результате наследственной ферментопатии. Эти дистрофии относятся к так называемым болезням накопления.
Наиболее яркими примерами наследственных дистрофий, связанных с нарушением внутриклеточного метаболизма аминокислот, являются цистиноз, тирозиноз, фенилпировиноградная олигофрения (фенилкетонурия). Их характеристика представлена в табл. 1.
Таблица 1.Наследственные дистрофии, связанные с нарушением обмена аминокислот
| Название | Дефицит фермента | Локализация накоплений аминокислоты |
| Цистиноз | Неизвестен | Печень, почки, селезенка, глаза, костный мозг, лимфатические узлы, кожа |
| Тирозиноз | Тирозинаминотрансфераза или оксидаза пара-оксифенилпировиноградной кислоты | Печень, почки, кости |
| Фенилпирови- ноградная олигофрения | Фенилаланин-4-гидроксилаза | Нервная система, мышцы, кожа, кровь, моча |
Паренхиматозные жировые дистрофии (липидозы)
В цитоплазме клеток содержатся в основном липиды, которые образуют с белками сложные лабильные жиробелковые комплексы - липопротеиды. Эти комплексы составляют основу мембран клетки. Липиды вместе с белками являются составной частью и клеточных ультраструктур. Помимо липопротеидов, в цитоплазме встречаются и нейтральные жиры, которые представляют собой сложные эфиры глицерина и жирных кислот.
Для выявления жиров используют срезы нефиксированных замороженных или фиксированных в формалине тканей. Гистохимически жиры выявляются с помощью ряда методов: судан III и шарлах окрашивают их в красный цвет, судан IV и осмиевая кислота - в черный, сульфат нильского голубого окрашивает жирные кислоты в темно-синий цвет, а нейтральные жиры - в красный.
С помощью поляризационного микроскопа можно дифференцировать изотропные и анизотропные липиды, последние дают характерное двойное лучепреломление.
Нарушения обмена цитоплазматических липидов могут проявляться в увеличении их содержания в клетках, где они обнаруживаются и в норме, в появлении липидов там, где они обычно не встречаются, и в образовании жиров необычного химического состава. Обычно в клетках накапливаются нейтральные жиры.
Паренхиматозная жировая дистрофия встречается наиболее часто там же, где и белковая, - в миокарде, печени, почках.
В миокардежировая дистрофия характеризуется появлением в мышечных клетках мельчайших жировых капель (пылевидное ожирение). При нарастании изменений эти капли (мелкокапельное ожирение) полностью замещают цитоплазму (рис. 29). Большинство митохондрий при этом распадается, поперечная исчерченность волокон исчезает. Процесс имеет очаговый характер и наблюдается в группах мышечных клеток, расположенных по ходу венозного колена капилляров и мелких вен.

Рис. 29.Жировая дистрофия миокарда:
а - капли жира (на рисунке черного цвета) в цитоплазме мышечных волокон (микроскопическая картина); б - включения липидов (Л), имеющие характерную исчерченность; Мф - миофибриллы. Электронограмма. х21 000
Внешний вид сердца зависит от степени жировой дистрофии. Если процесс выражен слабо, его можно распознать лишь под микроскопом, применяя специальные окраски на липиды; если он выражен сильно, сердце выглядит увеличенным в объеме, камеры его растянуты, оно дряблой консистенции, миокард на разрезе тусклый, глинисто-желтый. Со стороны эндокарда видна желто-белая исчерченность, особенно хорошо выраженная в сосочковых мышцах и трабекулах желудочков сердца («тигровое сердце»). Эта исчерченность миокарда связана с очаговым характером дистрофии, преимущественным поражением мышечных клеток вокруг венул и вен. Жировая дистрофия миокарда рассматривается как морфологический эквивалент его декомпенсации.
Развитие жировой дистрофии миокарда связывают с тремя механизмами: повышенным поступлением жирных кислот в кардиомиоциты, нарушением обмена жиров в этих клетках и распадом липопротеидных комплексов внутриклеточных структур. Чаще всего эти механизмы реализуются путем инфильтрации и декомпозиции (фанероза) при энергетическом дефиците миокарда, связанном с гипоксией и интоксикацией (дифтерия). При этом основное значение декомпозиции не в высвобождении липидов из липопротеидных комплексов клеточных мембран, а в деструкции митохондрий, что ведет к нарушению окисления жирных кислот в клетке.
В печенижировая дистрофия (ожирение) проявляется резким увеличением содержания жиров в гепатоцитах и изменением их состава. В клетках печени вначале появляются гранулы липидов (пылевидное ожирение), затем мелкие капли их (мелкокапельное ожирение), которые в дальнейшем
сливаются в крупные капли (крупнокапельное ожирение) или в одну жировую вакуоль, которая заполняет всю цитоплазму и отодвигает ядро на периферию. Измененные таким образом печеночные клетки напоминают жировые. Чаще отложение жиров в печени начинается на периферии, реже - в центре долек; при значительно выраженной дистрофии ожирение клеток печени имеет диффузный характер.
Внешний вид печени достаточно характерен: она увеличена, дряблая, охряно-желтого или желто-коричневого цвета. При разрезе на лезвии ножа и поверхности разреза виден налет жира.
Среди механизмов развития жировой дистрофии печени различают: чрезмерное поступление в гепатоциты жирных кислот или повышенный их синтез этими клетками; воздействие токсических веществ, блокирующих окисление жирных кислот и синтез липопротеидов в гепатоцитах; недостаточное поступление в печеночные клетки аминокислот, необходимых для синтеза фосфолипидов и липопротеидов. Из этого следует, что жировая дистрофия печени развивается при липопротеидемии (алкоголизм, сахарный диабет, общее ожирение, гормональные расстройства), гепатотропных интоксикациях (этанол, фосфор, хлороформ и др.), нарушениях питания (недостаток белка в пище - алипотропная жировая дистрофия печени, авитаминозы, болезни пищеварительной системы).
В почкахпри жировой дистрофии жиры появляются в эпителии проксимальных и дистальных канальцев. Обычно это нейтральные жиры, фосфолипиды или холестерин, который обнаруживают не только в эпителии канальцев, но и в строме. Нейтральные жиры в эпителии узкого сегмента и собирательных трубок встречаются как физиологическое явление.
Внешний вид почек: они увеличены, дряблые (при сочетании с амилоидозом плотные), корковое вещество набухшее, серое с желтым крапом, заметным на поверхности и разрезе.
Механизм развития жировой дистрофии почек связан с инфильтрацией эпителия почечных канальцев жиром при липемии и гиперхолестеринемии (нефротический синдром), что ведет к гибели нефроцитов.
Причиныжировой дистрофии разнообразны. Чаще всего она связана с кислородным голоданием (тканевая гипоксия), поэтому жировая дистрофия так часто встречается при заболеваниях сердечно-сосудистой системы, хронических заболеваниях легких, анемиях, хроническом алкоголизме и т.д. В условиях гипоксии страдают в первую очередь отделы органа, находящиеся в функциональном напряжении. Вторая причина - инфекции (дифтерия, туберкулез, сепсис) и интоксикации (фосфор, мышьяк, хлороформ), ведущие к нарушениям обмена (диспротеиноз, гипопротеинемия, гиперхолестеринемия), третья - авитаминозы и одностороннее (с недостаточным содержанием белков) питание, сопровождающееся дефицитом ферментов и липотропных факторов, которые необходимы для нормального жирового обмена клетки.
Исходжировой дистрофии зависит от ее степени. Если она не сопровождается грубым поломом клеточных структур, то, как правило, оказывается обратимой. Глубокое нарушение обмена клеточных липидов в
большинстве случаев заканчивается гибелью клетки, функция органов при этом резко нарушается, а в ряде случаев и выпадает.
Группу наследственных липидозов составляют так называемые системные липидозы, возникающие вследствие наследственного дефицита ферментов, участвующих в метаболизме определенных липидов. Поэтому системные липидозы относят к наследственным ферментопатиям (болезни накопления), поскольку дефицит фермента определяет накопление субстрата, т.е. липидов, в клетках.
В зависимости от вида накапливающихся в клетках липидов различают: цереброзидлипидоз, илиглюкозилцерамидлипидоз (болезнь Гоше), сфингомиелинлипидоз (болезнь Ниманна-Пика),ганглиозидлипидоз (болезнь Тея-Сакса, или амавротическая идиотия), генерализованный ганглиозидоз(болезнь Нормана-Ландинга) и др. Чаще всего липиды накапливаются в печени, селезенке, костном мозге, центральной нервной системе (ЦНС), нервных сплетениях. При этом появляются характерные для того или иного вида липидоза клетки (клетки Гоше, клетки Пика), что имеет диагностическое значение при изучении биоптатов (табл. 2).
Таблица 2.Системные липидозы (наследственные ферментопатии, болезни накопления, лизосомные болезни)
| Название | Дефицит фермента | Локализация накоплений липида | Диагностический критерий при биопсии |
| Болезнь Гоше - цереброзидлипидоз или глюкозидцерамидлипидоз | Глюкоцереброзидаза | Печень, селезенка, костный мозг, ЦНС (у детей) | Клетки Гоше |
| Болезнь Ниманна- Пика - сфингомиелинлипидоз | Сфингомиелиназа | Печень, селезенка, костный мозг, ЦНС | Клетки Пика |
| Амавротическая идиотия, болезнь Тея-Сакса - ганглиозидлипидоз | Гексозаминидаза | ЦНС, сетчатка глаз, нервные сплетения, селезенка, печень | Изменения мейсснеровского (ректобиопсия) |
| Болезнь Нормана- Ландинга - генерализованный ганглиозидоз | -Галактозидаза | ЦНС, нервные сплетения, печень, селезенка, костный мозг, почки и др. | Отсутствует |
Шок
Шок (от франц. choc) - остро развивающийся патологический процесс, обусловленный действием сверхсильного раздражителя и характеризующийся нарушением деятельности ЦНС, обмена веществ и главное ауторегуляции микроциркуляторной системы, что ведет к деструктивным изменениям органов и тканей.
В основе шока различного происхождения лежит единый сложный многофазный механизм развития. Но для раннего периода шока характерны относительно специфичные признаки, обусловленные особенностями этиологии и патогенеза.
Исходя из этого, различают следующие виды шока: 1) гиповолемический, в основе которого лежит острое уменьшение объема циркулирующей крови (или жидкости); 2) травматический, пусковым механизмом которого является чрезмерная афферентная (преимущественно болевая) импульсация; 3) кардиогенный, возникающий в результате быстрого падения сократительной функции миокарда и нарастания потока афферентной (преимущественно «гипоксической») импульсации; 4) септический (токсико-инфекционный), вызываемый эндотоксинами патогенной микрофлоры.
В поздний период шока относительная специфичность признаков, обусловленных особенностями его этиологии и патогенеза, исчезает, его клинико-морфологические проявления становятся стереотипными. 161
Для морфологической картины шока характерны нарушения гемокоагуляции в виде ДВС-синдрома, геморрагического диатеза, жидкой трупной крови, которые могут явиться основой для диагностики шока на вскрытии (Пермяков Н.К., 1979). Микроскопически нарушения гемодинамики и реологических свойств крови представлены распространенным спазмом сосудов, микротромбами в системе микроциркуляции, признаками повышенной проницаемости капилляров, геморрагиями. Во внутренних органах развивается ряд общих изменений в виде дистрофии и некроза, обусловленных нарушениями гемодинамики, гипоксией, повреждающим действием биогенных аминов, эндотоксинов патогенной микрофлоры. Выраженность этих изменений в значительной мере определяет возможность обратимости шока.
Морфологические изменения при шоке могут иметь ряд особенностей, обусловленных как структурно-функциональной специализацией органа, так и преобладанием в патогенезе шока одного из его звеньев - нейрорефлекторного, гипоксического, токсического.
Руководствуясь этим положением, при характеристике шока стали использовать термин «шоковый орган».
В шоковой почке тяжелым дистрофическим и некротическим изменениям подвергаются наиболее функционально отягощенные отделы нефрона - проксимальные канальцы; развивается некротический нефроз (иногда симметричные кортикальные некрозы почек), что обусловливает острую почечную недостаточность при шоке. В шоковой печени гепатоциты теряют гликоген, подвергаются гидропической дистрофии, развиваются центролобулярные некрозы печени, появляются признаки структурнофункциональной недостаточности звездчатых ретикулоэндотелиоцитов. Все эти изменения определяют возможность развития острой печеночной недостаточности при шоке. При этом нередко сочетание почечной и печеночной недостаточности, тогда говорят о гепаторенальном синдроме.
Шоковое легкое характеризуется очагами ателектаза, серозногеморрагическим отеком с выпадением фибрина в просвет альвеол, гемостазом и тромбами в микроциркуляторном русле, что обусловливает развитие острой дыхательной недостаточности.
Структурные изменения миокарда при шоке представлены дистрофическими и некробиотическими изменениями кардиомицитов: исчезновением гликогена, появлением липидов и контрактур миофибрилл. Возможно появление мелких очагов некроза.
Выраженные структурные повреждения при шоке выявляются не только в шоковых органах, но и в желудочно-кишечном тракте, нервной, эндокринной и иммунной системах.