Наследственные дисплазии соединительной ткани
Аутосомно-доминантные заболевания
При аутосомно-доминантном наследовании гетерозиготное носительство мутации оказывается достаточным для проявления заболевания. При этом мальчики и девочки болеют с одинаковой частотой. В количественном отношении доминантных заболеваний больше, чем рецессивных. В отличие от рецессивных доминантные мутации не приводят к полной инактивации функции кодируемого белка. Их эффект обусловлен либо снижением его количества (так называемая гаплонедоста-точность), либо появлением у мутантного белка нового агрессивного свойства.
Вероятность рождения больных детей в браке гетерозиготного носителя доминантной мутации со здоровым супругом составляет 50%. Поэтому аутосомно-доминантные заболевания могут носить семейный характер и передаваться из поколения в поколение, причем среди родственников только со стороны одного из родителей больного. Такой тип передачи заболевания иногда называют наследование «по вертикали». Если оба родителя ребенка с доминантным заболеванием оказываются здоровыми, можно предположить, что болезнь развилась вследствие возникновения новой мутации в половых клетках одного из супругов.
По некоторым данным около 80% синдромов с аутосомно-доминантным типом наследования являются следствием мутаций de novo в половых клетках отцов. В этом случае риск повторного рождения больного ребенка такой же, как в любых других семьях. Исключением из этого правила являются доминантные заболевания с неполным проявлением или неполной пенетрантностью, когда на развитие заболевания дополнительно оказывают влияния какие-то внешние факторы или чаще состояния каких-то других генов. В этих случаях носители доминантной мутации могут быть здоровыми, а их дети больными или наоборот. Пенетрант-ность выше 60% является высокой степенью повторяемости заболевания в поколениях. Доминантный ген может обладать разной экспрессивностью, то есть внутри одной семьи картина заболевания может варьировать по степени тяжести и клиническим проявлениям. Напомним, что термины пенетрантность и экспрессивность в генетическую практику были введены известным отечественным генетиком Н. В.Тимофеевым-Ресовским. Об этом крупном ученом генетике и интереснейшем человеке петербургский писатель Даниил Гранин написал повесть «Зубр».
Доминантные мутации в гомозиготном состоянии у больных встречаются редко, и, как правило, они ассоциированы с более тяжелой клиникой. Так, при гетерозиготном носительстве доминантной мутации в гене рецептора липопротеинов низкой плотности у больных семейной гиперхолестеринемией ишемическая болезнь сердца и инфаркт миокарда развиваются в возрасте 30-40 лет, тогда как при гомозиготном носительстве - в первой декаде жизни. При доминантном типе наследования не происходит накопления мутаций в популяции, так как больные часто не оставляют потомства в силу тяжести своего состояния. Многие доминантные заболевания проявляются в достаточно позднем возрасте.В конце прошлого века было показано, что самыми распространёнными аутосомно-доминантными заболеваниями являются наследственные опухолевые синдромы. Их суммарная частота в различных популяциях достигает 1%, причем чаще всего обусловливающие их мутации передаются из поколения в поколение, а не возникают de novo.
Наследственные дисплазии соединительной ткани
Наследственные дисплазии соединительной ткани - это гетерогенная группа моногенных болезней, обусловленных присутствием мутаций в генах белков внеклеточного матрикса или ферментов их биосинтеза, а также в генах, участвующих в регуляции морфогенеза соединительной ткани. Большинство этих заболеваний наследуются по аутосомно-доминантному типу. Ведущая роль в поддержании структурной целостности различных соединительных тканей принадлежит коллагенам, большому семейству близкородственных внеклеточных матриксных белков, составляющему более 30% общей массы белков тела млекопитающих. Открытие около 40 коллагеновых генов и расшифровка их молекулярной природы создали предпосылки для изучения молекулярных основ этиологии и патогенеза наследственных коллагенопатий - гетерогенной группы из более чем 70 моногенных заболеваний.
Наиболее известным генетическим вариантом наследственной дисплазии соединительной ткани является синдром Марфана. Долгое время предполагали, что это заболевание обусловлено мутациями в одном из коллагеновых генов. Однако оказалось, что при синдроме Марфана первичным биохимическим дефектом является нарушение структуры фибриллина 1 - структурного белка микрофибриллярных эластических волокон внеклеточного матрикса. Наряду с этим, описаны другие аутосомно-доминантные заболевания, при которых у больных наблюдается марфаноподобный фенотип. Остановимся более подробно на этих двух группах наследственных дисплазии соединительной ткани.
Наследственные коллагенопатии
В настоящее время известно 27 различных типов коллагеновых белков. Каждый из них состоит из трех равномерно скрученных полипептидных альфа-цепей, образующих структуру, подобную трехгранному шнуру. Разные типы коллагенов могут быть образованы либо тремя одинаковыми альфа-цепями, либо двумя или тремя различными полипептидами в соотношении 2:1 или 1:1:1 соответственно. Каждая альфа-цепь кодируется собственным геном, поэтому разнообразие коллагеновых генов больше, чем разнообразие соответствующих белков. Биосинтез зрелых коллагенов сопровождается необычно большим числом пост трансляционных модификаций, так что на одной молекуле проколлагеновой полипептидной цепи осуществляется более 120 реакций. В этих превращениях принимают участие более десятка различных ферментов. Все зрелые коллагеновые белки способны к образованию крупных су-прамолекулярных агрегатов. На рис.45 показаны основные этапы биосинтеза коллагена.
Любая альфа-цепь содержит коллагеновой домен, на всем протяжении которого за исключением короткого С-терминального участка каждая третья аминокислота является глицином. Таким образом, молекулярная формула коллагенового домена может быть записана как (Gly-X-Y)n, где X и Y - аминокислоты не-Gly типа. Различные коллагеновые альфа-цепи различаются по количеству и протяженности (Gly-X-Y)-MOTHBOB в коллагеновом домене и по конкретному содержанию аминокислот в X и Y положениях. Присутствие глицина, самой маленькой из аминокислот, в каждом третьем положении коллагеновых полипептидных цепей существенно для их правильного скручивания в тройную спираль, так как глицин при этом занимает ограниченное пространство в центре триплекса. Поэтому любые мутации, приводящие к замене глицина на другую аминокислоту, будут сопровождаться локальными нарушениями структуры тройной спирали и дезорганизацией более крупных агрегатов коллагена. К тяжелым последствиям также приводят мутации, нарушающие структуру С-концевого участка адьфа-цепи, так как образование триплекса по типу «застежки-молнии» начинается именно с этого участка молекулы. Кроме того, именно в этой области локализованы сайты взаимодействия коллагена более чем с 50 другими белками. Патологический процесс оказывается менее тяжелым, если в результате мутации альфа-цепь полностью утрачивает способность участвовать в формировании зрелых коллагеновых молекул. Это мутации, сопровождающиеся преждевременной терминацией трансляции или затрагивающие N-концевые районы альфа-цепи коллагена. При этом в образовании триплексной структуры принимают участие только нормальные полипептиды, мутантные альфа-цепи в нее не входят и вскоре после синтеза подвергаются внутриклеточному протеолизу. В результате снижается скорость синтеза зрелых коллагеновых молекул, но их структура сохраняется нормальной, и они не утрачивают способность к образованию упорядоченных супрамолекулярных агрегатов. Доминантный характер заболеваний, обусловленных нарушением структуры коллагеновых молекул, объясняется тем, что присутствие, наряду с мутантными, нормальных альфа-цепей не предотвращает образования дефектов в фибриллах или других надмолекулярных комплексах коллагена. Заболевания, вызванные нарушением биосинтеза коллагеновых молекул и связанные с присутствием мутаций в генах соответствующих ферментов, наследуются по рецессивному типу.
Коллагены I, II и III типов являются мажорными и составляют более 90% всех коллагенновых белков. Они способны формировать крупные высоко организованные фибриллы, в которых отдельные молекулы коллагена располагаются четырехступенчатыми уступами. Остальные коллагеновые белки относятся к классу нефибриллярных коллагенов, формирующих мелкие фибриллы, либо листовидные мембранные образования.
Коллаген I типа экспрессируется повсеместно, но особенно обильно представлен в костной системе, сухожилиях и коже. Коллаген II типа является мажорным хрящевым коллагеном. Он также составляет основу стекловидного тела. Кроме того, в хрящевой ткани экспрессируются минорные коллагены IX, X, XI и XII типов. Эмбриональный мажорный коллаген III типа является основным компонентом стенок сосудов и кишечника. В базальных мембранах присутствует коллаген IV типа. V коллаген образует каркас внутри фибрилл мажорных коллагенов. Коллаген VI типа участвует во взаимодействии между фибриллами мажорных коллагенов и другими структурными компонентами внеклеточного матрикса. Коллагены VII и XVII типов присутствуют в эпидермальных кератиноцитах и являются компонентами кожных опорных фибрилл. Коллагены VIII и XVIII типов найдены в эндотелии сосудов и роговице, они участвуют в регуляции неоваскуляризации и образовании мембраны Десцемета. Остальные коллагены ассоциируются с мажорными коллагенами I и II типов, способствуя их взаимодействию с другими белками внеклеточного матрикса. Очевидно, что структурные дефекты коллагенов могут сопровождаться тяжелыми повреждениями соединительной ткани. В настоящее время мутации, ассоциированные с различными нозологическими формами наследственных коллагенопатий, найдены в 25 коллагеновых генах, участвующих в синтезе 13 различных типов коллагенов. Клинические проявления этих заболеваний хорошо коррелируют с характером экспрессии различных типов коллагенов и с исполняемыми ими функциями.
Синдром Элерса-Данло
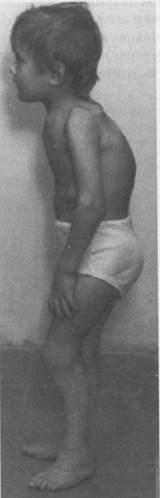
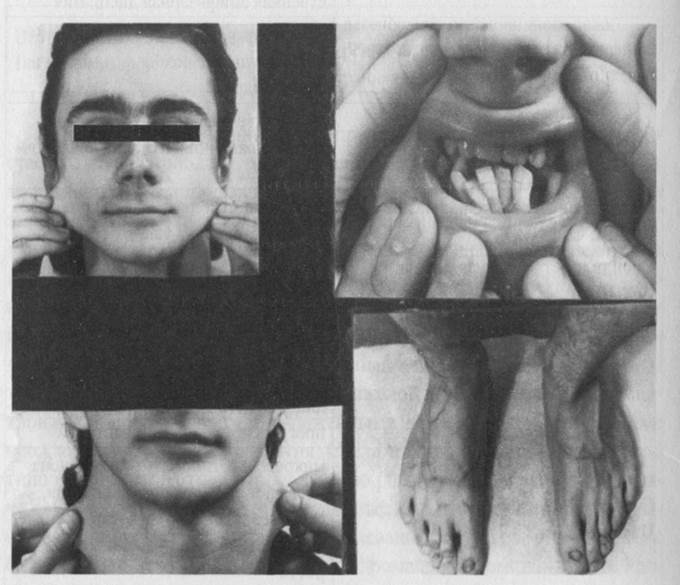
Классические варианты синдрома Элерса-Данло, характеризующиеся гиперрастяжимостью и истончением кожи, гипермобильностью суставов, неровным ростом зубов, деформацией ногтей, скелетными аномалиями, пролабированием клапанов сердца и др. клиническими проявлениями, обусловлены дефектами коллагена V типа.
|
|
Рисунок 1. Больной с синдромом Элерса-Данло
Наиболее тяжелым является «артериальный» тип заболевания, так как может сопровождаться разрывами артерий и перфорацией внутренних органов. При этом дефектным оказывается коллаген III типа, обильно представленный в стенках сосудов и кишечника. При VII типе синдрома Элерса-Данло, характеризующимся сверх гиперрастяжимостью и лёгкой ранимостью кожи, выраженной гипермобильностью суставов, нанизмом и скелетными дисплазиями, найдены специфические мутации в генах COL1A1 и COL1A2 коллагена I типа. Все мутации, идентифицированные у больных с данным типом заболевания затрагивают сайт узнавания для одной из протеаз, участвующих в процессинга коллагена I, а именно в удалении N-концевого пропептида. Остальные варианты синдрома Элерса-Данло наследуются по аутосомно-рецессивному типу, так как большинство из них обусловлено мутациями в генах ферментов биосинтеза коллагена.
Сопутствующими симптомами многих вариантов наследственных коллагенопатий и в первую очередь синдрома Элерса-Данло, являются дистрофия ногтей, несовершенный дентиногенез, парадонтоз.
Пролапс митрального и других клапанов сердца также может сопровождать наследственные коллагенопатий. В частности этот симптом входит в структуру синдрома Стиклера и классических форм синдрома Элерса-Данло.