Условия проведения ферментативной реакции.
Глава 4. ФЕРМЕНТАТИВНАЯ КИНЕТИКА
4.1. Особенности организации и функционирования ферментов. Закономерности ферментативного катализа
Реакции, протекающие в живых организмах, подчиняются общим законам химии, однако отличаются высокой специфичностью и отсутствием побочных продуктов. Эта особенность определяется свойствами ферментов. Ферменты (энзимы) – это специфические высокоэффективные белковые катализаторы химических реакций. Большинство клеточных реакций осуществляется с участием ферментов. Обмен веществ в клетках был бы невозможен без резкого ускорения химических реакций, без согласования во времени и пространстве множества биохимических процессов, т. е. без участия ферментов. Одна клетка может содержать до 1000 различных ферментов. В настоящее время известны функции более 2000 ферментов, из которых для нескольких сотен определена аминокислотная последовательность и пространственная структура.
Свойства ферментов.Как и другие химические катализаторы, ферменты:
· увеличивают скорость реакции, но не расходуются в ходе процесса и не претерпевают необратимых изменений;
· не смещают равновесие химической реакции, ускоряя как прямую, так и обратную реакцию в равной степени;
· повышают скорость реакции, снижая энергию активации, т. е. тот энергетический барьер, который требуется преодолеть для осуществления реакции.
Ферменты отличаются от химических катализаторов следующими свойствами:
1) высокой эффективностью действия – ферментативный катализ ускоряет протекание химических реакций в 106−1014 раз;
2) высокой специфичностью действия – способностью связываться с определенным субстратом и катализировать реакцию определенного типа;
3) «мягкими» условиями протекания ферментативных реакций – нормальное атмосферное давление, температура 30−40°С, рН ~ 7, водная среда;
4) способностью к регуляции своей активности, которая позволяет клеткам четко координировать осуществление многочисленных разветвленных метаболических реакций, обеспечивая наиболее высокий и экономный уровень обмена веществ, а также быструю приспособляемость к меняющимся условиям окружающей среды.
Классификация ферментов. Поскольку для ферментов характерна специфичность действия, их классифицируют по типу реакции, подвергающейся катализу. Согласно принятой в настоящее время классификации, ферменты группируют в 6 классов.
1. Оксидоредуктазы (окислительно-восстановительные реакции):
Авосст + Вокисл → Аокисл + Ввосст
2. Трансферазы (реакции переноса функциональных групп между субстратами):
А−Х + В → А + В−Х
3. Гидролазы (реакции гидролиза, акцептором переносимой группы является молекула воды):
А−В + Н2О → А−Н + В−ОН
4. Лиазы (реакции отщепления групп от субстрата негидролитическим путем с образованием двойной связи или присоединения групп по двойным связям):
А(ХН)−В → А−Х + В−Н
5. Изомеразы (реакции изомеризации):
А ⇄ Изо-А
6. Лигазы или синтетазы (реакции синтеза за счет энергии расщепления нуклеозидтрифосфатов, чаще АТФ):
А + В + АТР → А−В + ADP + Рi
Номер соответствующего класса фермента закреплен в его кодовой нумерации (шифре). Шифр фермента состоит из четырех разделенных точками чисел, обозначающих класс фермента, подкласс, подподкласс и порядковый номер в подподклассе.
Систематические названия ферментов образуются путем добавления суффикса -аза к названию субстрата, на который воздействует данный фермент (в случае бимолекулярной реакции – к названиям двух субстратов, разделенных знаком деления), либо к названию типа катализируемой реакции. Например, аргиназа (катализирует гидролиз аргинина), алкогольдегидрогеназа (катализирует окисление этанола). После названия фермента в скобках указывают название органа или организма, из которого был выделен данный фермент. Например, алкогольдегидрогеназа (дрожжи) или алкогольдегидрогеназа (печень крыс).
В некоторых случаях до сих пор сохраняются тривиальные названия ферментов с окончанием -ин, не несущие химической информации, например пепсин и трипсин (протеолитические ферменты), каталаза (разрушает перекись водорода) и др.
Систематические названия ферментов используются тогда, когда необходима точная идентификация фермента. Многие систематические названия очень громоздки и удобнее пользоваться тривиальными названиями. Например, гексокиназа (тривиальное название) – это АТФ:D-гексозо-6-фосфотрансфераза.
Особенности структуры ферментов.Молекулы ферментов характеризуются молекулярными массами от 10 до 1000 кДа и выше, однако большинство ферментов представлено глобулярными белками с молекулярной массой в несколько сотен тысяч дальтон, построенными из субъединиц – протомеров. Ферменты функционируют обычно в составе мультиферментных систем, катализирующих определенные последовательности реакций (продукт реакции, полученный при участии одного фермента, является субстратом для второго фермента и т. д.). Упаковка субъединиц в мультимерном (состоящем из нескольких субъединиц) белке осуществляется благодаря взаимодействиям того же типа, что и при образовании четвертичной структуры белка. Среди ферментов-мультимеров преобладают димеры и тетрамеры, менее распространены гекса- и октамеры и очень редко встречаются тримеры и пентамеры. Например, дрожжевая синтетаза жирных кислот, катализирующая синтез жирных кислот из низкомолекулярных предшественников, представляет собой систему из семи разных ферментов, молекулы которых объединены в прочно связанный комплекс.
Мультимерные ферментные белки могут содержать протомеры нескольких типов, катализирующих одну и ту же реакцию, но различающихся первичной структурой, молекулярной массой, субстратной специфичностью и др. От соотношения протомеров разного типа в мультимере зависят некоторые его физические и химические свойства. Такие различающиеся формы мультимерного фермента называются изоферментами(изозимами).
Изоферменты являются продуктами экспрессии разных генов. В виде нескольких изоферментов существует ряд ферментов, причем они могут встречаться у одного и того же организма и даже внутри одной и той же клетки. Один из основных механизмов образования изоферментов включает объединение разных субъединиц в разной комбинации при образовании активного олигомерного фермента. Например, лактатдегидрогеназа, катализирующая в мышцах обратимую реакцию окисления молочной кислоты, состоит из четырех субъединиц (тетрамер) двух типов (Н и М) и представлена пятью изоферментами – НННН, НННМ, ННММ, НМММ, ММММ. Они отличаются друг от друга активностью, молекулярной массой, электрофоретической подвижностью, локализацией в органах и тканях, чувствительностью к регуляторным веществам. Существование изоферментов позволяет организму изменять их соотношение и регулировать таким образом метаболическую активность.
Изучение структуры молекул ферментов дало возможность выявить ряд закономерностей в их организации. Полипептидная цепь, образующая белковую глобулу, свернута довольно сложным образом. Одни участки этой цепи являются a-спиралями или же b-структурами, другие принимают нерегулярные, но вполне определенные конформации. Эти структуры, тесно прилегая друг к другу и чередуясь, упаковываются в блоки, обладающие функциональной активностью. На поверхности белковой глобулы находятся в основном полярные группы и заряженные атомы, причем между противоположно заряженными группами иногда образуются ионные связи. Внутренние области белковой глобулы представляют собой неполярную среду, гидрофобное ядро образовано неполярными группами, входящими главным образом в состав алифатических и ароматических боковых цепей аланина, валина, лейцина, изолейцина, метионина, фенилаланина и триптофана. Полярные радикалы аминокислот, имеющие функциональное значение, могут быть также ориентированы внутрь глобулы и ассоциированы друг с другом.
Важнейшей частью фермента является активный центр, обычно имеющий форму щели или впадины в глобуле фермента и представляющий собой сложную трехмерную структуру (рис. 4.1). Одни ферменты имеют один, другие – два или более активных центров. В активном центре происходит связывание субстрата и превращение его в продукт. Активный центр почти всегда построен из небольшого количества аминокислотных остатков, которые, как правило, значительно удалены друг от друга в полипептидной цепи. При ее свертывании функциональные группы этих аминокислотных остатков сближаются и формируют активный центр.
Рис. 4.1. Связывание субстрата в активном центре фермента
В активном центре выделяют два участка – связывающий и каталитический. Остатки аминокислот, образующие связывающий участок, отвечают за специфическое комплементарное связывание субстрата и образование фермент-субстратного комплекса, обеспечивая удержание субстрата в активном центре. Именно «архитектура» связывающего участка активного центра фермента определяет его комплементарность структуре субстрата. Формирование фермент-субстратного комплекса происходит без образования ковалентных связей, за счет более слабых сил – водородных и электростатических связей, гидрофобных и ван-дер-ваальсовых взаимодействий.
В каталитический участок фермента входят остатки аминокислот, непосредственно участвующие в катализе. Их называют каталитическими группами, и они чаще всего представлены функциональными группами остатков серина, гистидина, триптофана, аргинина, цистеина, аспарагиновой и глутаминовой кислоты, тирозина. Окончательное формирование каталитического участка у многих ферментов может происходить в момент присоединения субстрата (принцип индуцированного соответствия субстрата и фермента).
Активный центр не может быть очерчен строго определенными границами, поскольку каждый его компонент так или иначе взаимодействует с другими участками молекулы фермента. Влияние микроокружения может быть весьма существенным:
· компоненты активного центра, в том числе и кофакторы, взаимодействуют с соседними группами фермента, что изменяет химические свойства функциональных групп, участвующих в катализе;
· в клетке ферменты образуют структурные комплексы как друг с другом, так и с участками клеточных и внутриклеточных мембран, с элементами цитоскелета и/или другими молекулами, что влияет на реакционную способность функциональных групп в активном центре фермента.
Структура активного центра определяет регио- и стереоспецифичность действия ферментов.
Некоторые ферменты проявляют полифункциональность – способность катализировать несколько типов реакций. Это явление объясняется тем, что при формировании третичной структуры полипептидные цепи таких ферментов образуют несколько функционально и стерически обособленных глобулярных участков – доменов, каждый из которых характеризуется собственной каталитической активностью.
Специфичность ферментов. Одним из удивительных свойств ферментов является их высокая специфичность. Различают субстратную специфичность и реакционную специфичность. Большинство ферментов высокоспецифично как к природе, так и к пути превращения субстрата.
Субстратная специфичность – это способность фермента катализировать превращение определенного субстрата или нескольких субстратов со схожей химической структурой. Эта специфичность у разных ферментов значительно варьирует: одни ферменты могут катализировать реакцию с участием только одного субстрата (абсолютная специфичность), другие взаимодействуют с несколькими химически родственными веществами (групповая специфичность). Например, формамидаза гидролизует только формамид, а амидаза – любой алифатический амид. В этом случае говорят соответственно об узкой и широкой субстратной специфичности ферментов.
Субстратная специфичность обусловлена комплементарностью структуры связывающего участка фермента структуре субстрата. Между аминокислотными остатками активного центра фермента и субстратом устанавливается геометрическое (по форме) и химическое соответствие (образование гидрофобных, ионных и водородных связей). Связывание субстрата в активном центре фермента происходит многоточечно, с участием нескольких функциональных групп.
В настоящее время получила развитие общепринятая гипотеза Кошланда об индуцированном соответствии субстрата и фермента. Согласно этой гипотезе, пространственное соответствие между структурами активного центра фермента и субстрата создается в момент их взаимодействия друг с другом. При этом в молекуле фермента индуцируются небольшие конформационные изменения, в результате чего в каталитическом участке функционально активные группы ориентируются наиболее благоприятным для протекания реакции образом. В субстрате также возникают конформационные изменения (говорят о возникновении напряжения в субстрате), что делает его более реакционноспособным. Конформационная подвижность крупной молекулы фермента создает, следовательно, дополнительные возможности для усиления его каталитической активности.
Данная гипотеза объясняет то, что молекулы, очень похожие по форме на истинный субстрат, могут связываться с ферментом, но не превращаться в продукт, т. е. действуют как ингибиторы. Кроме того, смысл индуцированных конформационных изменений фермента состоит в том, чтобы обеспечить освобождение продукта(ов) реакции из активного центра, после чего конформация молекулы фермента возвращается в исходное состояние.
Реакционная специфичностьхарактеризует способность ферментов катализировать реакции определенного типа (например, окислительно-восстановительные). Если субстрат может существовать в нескольких изомерных формах, то одни и те же химические превращения этих изомеров катализируют разные ферменты (например, оксидазы L-аминокислот и оксидазы D-аминокислот). Исключение составляют изомеразы, которые катализируют взаимопревращения изомеров.
Закономерности ферментативного катализа. Ферментативная реакция – это многостадийный процесс. На первой стадии устанавливается индуцированное комплементарное соответствие между ферментом (Е) и субстратом (S). В результате образуется фермент-субстратный комплекс (ЕS), в котором далее происходит химическое превращение субстрата в продукт(ы). ЕS-комплекс через переходное состояние ЕS* превращается в комплекс фермент-продукт(ы) (ЕР), после чего продукт(ы) превращения отделяется от фермента:
Е + S ⇄ ЕS ⇄ ЕS* ⇄ ЕР ⇄ Е + Р
При связывании субстрата с ферментом происходит изменение конформации молекул фермента и субстрата, последняя фиксируется в активном центре в напряженной конфигурации. Так формируется активированный комплекс, или переходное состояние – высокоэнергетическая промежуточная структура, которая энергетически менее устойчива, чем исходные соединения и продукты (рис. 4.2). Важнейший вклад в суммарный каталитический эффект вносит процесс стабилизации переходного состояния – взаимодействия между аминокислотными остатками белка и субстратом. Разность значений свободной энергии для исходных реагентов и переходного состояния соответствует свободной энергии активации(DG#). Это количество энергии, необходимое для перевода всех молекул субстрата в активированное состояние. Скорость реакции зависит от величины DG#: чем она меньше, тем больше скорость реакции, и наоборот. По сути DG# представляет собой энергетический барьер, который требуется преодолеть молекулам субстрата для осуществления реакции. Вершина энергетического барьера соответствует переходному состоянию. Стабилизация переходного состояния понижает этот барьер или энергию активации, т.е. ферменты повышают скорость реакций путем снижения активационного барьера и увеличения энергии субстрата при связывании его с ферментом, не влияя при этом на полное изменение свободной энергии (рис. 4.2).
Рис. 4.2. Модели превращения субстрата:
а – неферментативное превращение; б, в – ферментативное превращение
Можно выделить несколько причин высокой каталитической активности ферментов, которые обеспечивают снижение энергетического барьера реакции.
1. Фермент может связывать молекулы реагирующих субстратов таким образом, что их реакционноспособные группы будут располагаться поблизости друг от друга и от каталитических групп фермента (эффект сближения).
2. При образовании фермент-субстратного комплекса достигаются фиксация субстрата и его оптимальная для разрыва и образования химических связей ориентация (эффект ориентации).
3. Связывание субстрата приводит к удалению его гидратной оболочки (существует для растворенных в воде веществ).
4. Эффект индуцированного соответствия субстрата и фермента.
5. Стабилизация переходного состояния.
6. Определенные группы в молекуле фермента (кофермента) могут обеспечивать кислотно-основной (перенос протонов в субстрате) и нуклеофильный катализ (формирование ковалентных связей между ферментом и субстратом, что ведет к образованию более реакционноспособных структур, чем субстрат). Последний характерен для ферментов, катализирующих реакции нуклеофильного замещения.
Кофакторы ферментов.Активность ряда ферментов зависит только от структуры самого белка. Однако во многих случаях (~40%) для осуществления катализа ферменты нуждаются в особых посредниках – кофакторах. Кофакторы – это низкомолекулярные соединения небелковой природы (ионы металлов, сложные органические соединения, в основном производные витаминов), которые функционируют на промежуточных стадиях ферментативной реакции (или цикла реакций), но не расходуются в ходе катализа. В большинстве случаев кофакторы регенерируются в неизменном виде по завершении каталитического акта.
Отделение кофактора от белка, обычно связанного с ним нековалентными связями, приводит к образованию неактивного апофермента. Каталитически активный комплекс апофермент – кофактор называется холоферментом.
Различные по химической природе кофакторы делят на две основные группы – коферменты и простетические группы. Коферменты непрочно (нековалентно) связаны с белком и при катализе отделяются от него (например, НАД+, КоА). Восстановление их исходной структуры (регенерация) после участия в катализе может катализироваться уже другим ферментом. Простетические группы прочно (ковалентно) связаны с апоферментом и при катализе не отделяются от него (например, гем в гемопротеинах, атомы металлов в металлопротеинах).
Каждый кофактор имеет определенную структуру, что делает его специфичным для соответствующего типа реакций. Для участия в реакции кофакторы должны быть связаны с ферментами. При этом комплементарное, точное размещение кофактора в активном центре фермента обеспечивает множество нековалентных контактов с ферментом.
Основные механизмы, согласно которым кофакторы принимают участие в катализе, следующие:
· выполняют функцию переносчиков между ферментами. Взаимодействуя с одним ферментом, переносчик акцептирует часть субстрата, мигрирует к другому ферменту и передает переносимую часть субстрату второго фермента, после чего высвобождается. Такой механизм типичен для большинства коферментов;
· выполняют роль внутриферментного переносчика, что характерно, в первую очередь, для простетических групп. Простетическая группа присоединяет часть молекулы субстрата и переносит ее на второй субстрат, связанный в активном центре того же фермента. В этом случае простетическую группу рассматривают как часть каталитического участка фермента;
· изменяют конформацию молекулы фермента, взаимодействуя с ней вне активного центра, что может индуцировать переход активного центра в каталитически активную конфигурацию;
· стабилизируют конформацию фермента, способствующую образованию каталитически активного состояния;
· выполняют функцию матрицы. Например, полимеразы нуклеиновых кислот нуждаются в «программе» – матрице, по которой строится новая молекула;
· играют роль промежуточных соединений. Иногда фермент может использовать в реакции молекулу кофактора, образуя из нее продукт, но при этом одновременно за счет субстрата образовать новую молекулу кофактора.
Обычно кофакторы выполняют функцию промежуточных переносчиков электронов, некоторых атомов или функциональных групп, которые в результате ферментативной реакции переносятся с одного соединения на другое. Наиболее распространены кофакторы, осуществляющие перенос восстановительных эквивалентов, фосфатных, ацильных и карбоксильных групп. Мы ограничимся рассмотрением структуры и механизма функционирования переносчиков восстановительных эквивалентов.
Под восстановительными эквивалентами подразумевают обычно атомы Н, электроны или гидрид-ионы. Поскольку их перенос осуществляется в ходе окислительно-восстановительных реакций, соответствующие переносчики называют окислительно-восстановительными кофакторами:
Е1 Е2
АН2 + Р ⇄ А + РН2 РН2 + В ⇄ Р + ВН2
Суммарная реакция: АН2 + В ⇄ А + ВН2
где А, В – окисленные субстраты; Р – переносчик; АН2, ВН2 – восстановленные субстраты; Е1, Е2 – ферменты (дегидрогеназы).
К ним относятся никотинамидные и флавиновые переносчики, цитохромы, хиноны, липоевая и аскорбиновая кислоты, глутатион. Наиболее распространены никотинамидные (НАД+ и НАДФ+) и флавиновые (ФАД и ФМН) коферменты.
Никотинамидные переносчики восстановительных эквивалентов.Ими являются никотинамидадениндинуклеотид (НАД+, или NAD+) и никотинамидадениндинуклеотидфосфат (НАДФ+, или NADP+).
Окисленные формы этих коферментов принято обозначать НАД+ и НАДФ+, подчеркивая присутствие избыточного положительного заряда на атоме азота пиридинового кольца.
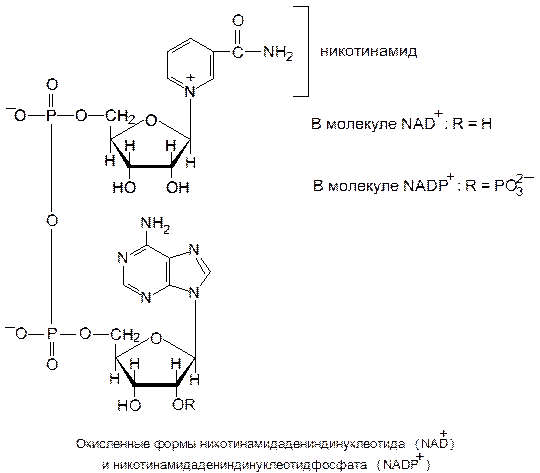
Функциональной группой никотинамидных переносчиков восстановительных эквивалентов служит пиридиновое кольцо, входящее в состав никотинамида – витамина В5 (РР). При ферментативном окислении субстрата с участием НАД+ (НАДФ+) никотинамид восстанавливается в ходе присоединения гидрид-иона Н− (Н− ⇄ Н+ + 2е−). При этом дегидрирование субстрата в большинстве случаев сопровождается отщеплением двух атомов водорода, в ходе которого протон (Н+) переносится через раствор.
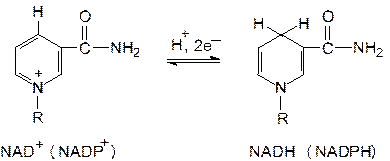
Примером функционирования никотинамидных переносчиков восстановительных эквивалентов может служить окисление этанола в уксусный альдегид, катализируемое алкогольдегидрогеназой (АДГ). Этот фермент осуществляет отщепление двух атомов водорода от молекулы этанола, причем к НАД+ переносится водород, связанный с углеродом спиртовой группы, а водород, присоединенный к кислороду ОН-группы, высвобождается в среду в виде протона.

Два пиридиновых кофермента участвуют в разных окислительно-восстановительных реакциях при различных окислительно-восстановительных потенциалах: НАД+ чаще выступает в роли окислительного агента в катаболитных путях, а НАДФ+ восстанавливается до НАДФН · Н+ и выполняет функцию восстановителя в биосинтетических процессах.
Флавиновые переносчики восстановительных эквивалентов. К ним относятся флавинадениндинуклеотид (ФАД, или FAD) и флавинмононуклеотид (ФМН, или FMN).
Реакционноспособной частью ФАД и ФМН служит изоаллоксазиновая система, содержащая двойные сопряженные связи. Структура этой системы изменяется при восстановлении. Дегидрирование с участием флавиновых кофакторов сопровождается отщеплением от субстрата двух атомов водорода, но в отличие от никотинамидных коферментов, акцептирующих гидрид-ион, флавиновые кофакторы акцептируют оба атома водорода. Поэтому восстановленные формы ФАД и ФМН обозначаются как ФАДН2 и ФМНН2.

Флавиновые коферменты являются более сильными окислителями, чем никотинамидные, а восстановленные формы никотинамидных коферментов служат более сильными восстановителями, чем восстановленные флавины.
4.2. Активность ферментов и факторы, на нее влияющие. Принципы ферментативной кинетики
Активность ферментов.Под активностью фермента понимают такое его количество, которое катализирует превращение определенного количества субстрата в единицу времени. Для выражения активности препаратов ферментов используют две альтернативные единицы: международную (МЕ) и катал (кат). За международную единицу активности фермента принято такое его количество, которое катализирует превращение 1 мкмоля субстрата в продукт за 1 мин в стандартных условиях (обычно оптимальных). Один катал обозначает количество фермента, катализирующее превращение 1 моля субстрата за 1 с (1 кат = 6 ∙ 107 МЕ). При бимолекулярной реакции A + В = С + D за единицу активности фермента принимают такое его количество, которое катализирует превращение 1 мкмоля А или В, или 2 мкмолей А (если В = А), за 1 мин.
Часто ферментные препараты характеризуются удельной активностью, которая отражает степень очистки фермента. Удельная активность – это число единиц активности фермента, приходящихся на 1 мг белка.
Молекулярная активность (число оборотов фермента) – число молекул субстрата, подвергающееся превращению одной молекулой фермента за 1 мин при полном насыщении фермента субстратом. Она равна числу единиц активности фермента, деленному на количество фермента, выраженное в микромолях. Понятие молекулярной активности применимо только для чистых ферментов.
Когда известно количество активных центров в молекуле фермента, вводится понятие активности каталитического центра. Она характеризуется числом молекул субстрата, которое подвергается превращению за 1 мин в расчете на один активный центр.
Активность ферментов сильно зависит от внешних условий, среди которых первостепенное значение имеют температура и рН среды. Повышение температуры в интервале 0−50°С обычно приводит к плавному увеличению ферментативной активности, что связано с ускорением процессов формирования фермент-субстратного комплекса и всех последующих событий катализа. При повышении температуры на каждые 10°С скорость реакции увеличивается примерно вдвое (правило Вант-Гоффа). Однако дальнейшее возрастание температуры (>50°С) сопровождается повышением количества инактивированного фермента за счет денатурации его белковой части, что выражается в снижении активности. Каждый фермент характеризуется температурным оптимумом – значением температуры, при котором регистрируется наибольшая его активность.
Зависимость активности ферментов от значения рН среды имеет сложный характер. Для каждого фермента характерен оптимум рНсреды, при котором он проявляет максимальную активность. При удалении от этого значения в ту или другую сторону ферментативная активность снижается. Это объясняется изменением состояния активного центра фермента (уменьшением или увеличением ионизации функциональных групп), а также третичной структуры всей белковой молекулы, которая зависит от соотношения в ней катионных и анионных центров. Большинство ферментов имеют оптимум рН в области нейтральных значений. Однако есть ферменты, проявляющие максимальную активность при рН 1,5 (пепсин) или 9,5 (аргиназа). При работе с ферментами необходимо поддерживать рН с помощью соответствующего буферного раствора.
Активность ферментов подвержена значительным колебаниям в зависимости от воздействия ингибиторов (веществ, частично или полностью снижающих активность) и активаторов (веществ, увеличивающих активность). Их роль выполняют катионы металлов, некоторые анионы, переносчики фосфатных групп, восстановительных эквивалентов, специфические белки, промежуточные и конечные продукты метаболизма и др.
Принципы ферментативной кинетики. Суть кинетических исследований состоит в определении максимальной скорости ферментативной реакции (Vmax) и константы Михаэлиса КМ. Ферментативная кинетика изучает скорости количественных превращений одних веществ в другие под действием ферментов. Скорость ферментативной реакции измеряют по убыли субстрата или приросту образующегося продукта за единицу времени, либо по изменению концентрации одной из смежных форм кофермента.
Влияние концентрации фермента на скорость реакции выражается в следующем: если концентрация субстрата постоянна (при условии избытка субстрата), то скорость реакции пропорциональна концентрации фермента. Для кинетических исследований используют концентрацию фермента 10-8 М активных центров. Оптимальное значение концентрации фермента определяют из графика зависимости активности фермента от его концентрации. Оптимальным считается значение, лежащее на плато полученного графика в области значений активности фермента, мало зависящих от его концентрации (рис. 4.3).
Рис. 4.3. Зависимость скорости ферментативной реакции
от концентрации фермента
Для изучения влияния концентрации субстрата на скорость ферментативной реакции сначала строят кинетическую кривую, отражающую изменение концентрации субстрата (S1) или продукта (Р1) во времени (рис. 4.4) и измеряют начальную скорость (V1) реакции как тангенс угла наклона касательной к кривой в нулевой точке.
Рис. 4.4. Кинетические кривые ферментативной реакции
Построив кинетические кривые для других значений концентрации данного субстрата (S2, S3, S4 и т. д.) или продукта (Р2, Р3, Р4 и т. д.) и определив начальные скорости (V2, V3, V4 и т. д.) реакции, строят график зависимости начальной скорости ферментативной реакции от концентрации субстрата (при постоянной концентрации фермента), который имеет вид гиперболы (рис. 4.5).
Рис. 4.5. Зависимость начальной скорости ферментативной реакции
от концентрации субстрата
Кинетика многих ферментативных реакций описывается уравнением Михаэлиса - Ментен. При постоянной концентрации фермента и малых значениях концентрации субстрата [S] начальная скорость реакции прямо пропорциональна [S] (рис. 4.5). В этом случае говорят о полунасыщении фермента субстратом, когда половина молекул фермента находится в форме фермент-субстратного комплекса и скорость реакции V = 1/2Vmax. В отношении субстрата реакция имеет 1-й порядок (скорость реакции прямо пропорциональна концентрации одного реагирующего вещества) или 2-й порядок (скорость реакции пропорциональна произведению концентраций двух реагирующих веществ).
При высоких значениях концентрации субстрата [S] скорость реакции почти не зависит от [S]: при дальнейшем увеличении [S] скорость реакции растет все медленнее и в итоге становится постоянной (максимальной) (рис. 4.5). При этом достигается полное насыщение фермента субстратом, когда все молекулы фермента находятся в форме фермент-субстратного комплекса и V = Vmax. В отношении субстрата реакция имеет 0-й порядок (скорость реакции не зависит от концентрации реагирующих веществ).
В 1913 г. Л. Михаэлис и М. Ментен предложили простую модель, объясняющую такую кинетику. Согласно этой модели, образование специфического фермент-субстратного комплекса является необходимым промежуточным этапом катализа.
k1 k3
Е + S ⇄ ЕS → Е + Р
k2
Фермент Е соединяется с субстратом S, образуя ЕS-комплекс. Константа скорости этого процесса k1. Судьба ЕS-комплекса складывается двояко: он может либо диссоциировать на фермент Е и субстрат S с константой скорости k2, либо подвергнуться дальнейшему превращению, образуя продукт Р и свободный фермент Е, с константой скорости k3. При этом постулируется, что продукт реакции не превращается в исходный субстрат. Это условие соблюдается на начальной стадии реакции, пока концентрация продукта невелика.
Скорость катализа определяют в стационарных условиях, когда концентрация промежуточных продуктов остается постоянной, тогда как концентрация исходных веществ и конечных продуктов изменяется. Это имеет место в том случае, когда скорость образования ЕS-комплекса равна скорости его распада.
Можно ввести новую константу КМ – константу Михаэлиса(моль/л), которая равна
 (4.1)
(4.1)
Уравнение Михаэлиса – Ментен, выражающее количественное соотношение между скоростью ферментативной реакции и концентрацией субстрата, имеет вид
 (4.2)
(4.2)
Это уравнение соответствует графику зависимости скорости реакции от концентрации субстрата. При низких концентрациях субстрата, когда [S] намного ниже КМ, V = Vmax[S] / КМ, т. е. скорость реакции прямо пропорциональна концентрации субстрата. При высоких концентрациях субстрата, когда [S] намного выше КМ, V = Vmax, т. е. скорость реакции максимальна и не зависит от концентрации субстрата.
Если [S] = КМ, то V = Vmax /2.
Таким образом, КМ равна концентрации субстрата, при которой скорость реакции составляет половину максимальной.
Константа Михаэлиса (КМ) и максимальная скорость реакции (Vmax) – важные характеристики скорости при разных концентрациях субстрата. Vmax – величина постоянная для каждого фермента позволяет оценить эффективность его действия.
Константа Михаэлиса показывает сродство субстрата к ферменту (в случае, когда k2 >> k3): чем меньше КМ, тем больше сродство и выше скорость реакции, и наоборот. Каждый субстрат характеризуется своей величиной КМ для данного фермента и по их значениям можно судить о субстратной специфичности фермента. Константа Михаэлиса зависит от природы субстрата, температуры, рН, ионной силы раствора и наличия ингибиторов.
В связи с тем, что определение Vmax и КМ непосредственно из графической зависимости Михаэлиса – Ментен (рис.4.5) является неоднозначным, прибегают к линеаризации данного уравнения. Для этого его преобразуют в такую форму, чтобы графически оно выражалось прямой. Существует несколько методов линеаризации, среди которых наиболее часто применяют методы Лайнуивера – Бэрка и Эди – Хофсти.
Преобразование Лайнуивера – Бэрка имеет вид
 (4.3)
(4.3)
Строят график зависимости 1/V = f (1/[S]) и получают прямую линию, пересечение которой с осью ординат дает величину 1/Vmax; отрезок, отсекаемый прямой на оси абсцисс дает величину −1/КМ, а тангенс угла наклона прямой к оси абсцисс равен КМ /Vmax (рис. 4.6). Этот график позволяет более точно определять Vmax. Как мы увидим ниже, из этого графика можно также извлечь ценную информацию, касающуюся ингибирования активности фермента.
Рис. 4.6. Метод линеаризации уравнения Михаэлиса – Ментен
(по Лайнуиверу – Бэрку)
Метод Эди – Хофсти основан на преобразовании уравнения Михаэлиса – Ментен путем умножения обеих его частей на Vmax:
 (4.4)
(4.4)
График в координатах V и V/[S] представляет собой прямую линию, пересечение которой с осью ординат дает величину Vmax, а отрезок, отсекаемый прямой на оси абсцисс, – величину Vmax/КМ (рис. 4.7). Он позволяет очень просто определять КМ и Vmax, а также выявлять возможные отклонения от линейности, не обнаруживаемые на предыдущем графике.
Рис. 4.7. Метод линеаризации уравнения Михаэлиса – Ментен
(по Эди – Хофсти)
Ингибирование активности ферментов. Действие ферментов можно полностью или частично подавить определенными химическими веществами – ингибиторами. По характеру своего действия ингибиторы подразделяются на обратимые и необратимые. В основе такого деления лежит прочность связывания ингибитора с ферментом.
Обратимые ингибиторы – это соединения, которые нековалентно взаимодействуют с ферментом и при их удалении активность фермента восстанавливается. Обратимое ингибирование может быть конкурентным, неконкурентным и бесконкурентным.
Примером конкурентного ингибирования является действие структурных аналогов субстрата, которые могут связываться с активным центром фермента похожим способом, как и субстрат, не превращаясь однако в продукт и препятствуя взаимодействию фермента с истинным субстратом, т. е. имеет место конкуренция между субстратом и ингбитором за связывание с активным центром фермента. В результате образования комплексов фермент-ингибитор (EI) концентрация ES-комплексов уменьшается и, как следствие, падает скорость реакции. Иными словами, конкурентный ингибитор уменьшает скорость катализа путем снижения доли молекул фермента, связавших субстрат.
Измерение скоростей реакций при разных концентрациях субстрата позволяет отличать конкурентное ингибирование от неконкурентного. При конкурентном ингибировании на графике зависимости 1/V = f (1/[S]) прямые пересекают ось ординат в одной точке 1/Vmax независимо от присутствия ингибитора, но в присутствии ингибитора увеличивается тангенс угла наклона прямой к оси абсцисс, т. е. Vmax не изменяется, а КМ увеличивается, что свидетельствует об уменьшении сродства субстрата к ферменту в присутствии ингибитора (рис. 4.8). Следовательно, при достаточно высокой концентрации субстрата в условиях конкуренции за активный центр фермента, когда субстрат вытесняет ингибитор из активного центра, ингибирование может быть устранено, и скорость катализируемой реакции восстанавливается. При этом уравнение Михаэлиса − Ментен имеет вид
 (4.5)
(4.5)
где [I] – концентрация ингибитора; Ki – константа ингибирования.
Константа ингибирования характеризует сродство фермента к ингибитору и представляет собой константу диссоциации ЕI-комплекса:
k1
Е + I ⇄ EI
k2

 (4.6)
(4.6)
В присутствии конкурентного ингибитора тангенс угла наклона прямой к оси абсцисс увеличивается на величину (1 + [I]/Ki).
Рис. 4.8. Конкурентное ингибирование:
а – схема; б – графическое выражение по Лайнуиверу − Бэрку
При неконкурентном ингибировании ингибитор отличается по структуре от субстрата и связывается не с активным, а с аллостерическим центром фермента. Это ведет к изменению конформации активного центра фермента, что сопровождается снижением каталитической активности фермента. Причем ингибитор может связываться не только со свободным ферментом (Е + I → EI), но и с фермент-субстратным комплексом (ES + I → ESI). Обе формы EI и ESI не активны. Субстрат и ингибитор могут быть одновременно связаны молекулой фермента, но участки их связывания не перекрываются. Действие неконкурентного ингибитора заключается в уменьшении числа оборотов фермента, а не в снижении доли связавших субстрат молекул фермента. Ингибитор не препятствует образованию ES-комплексов, но тормозит превращение субстрата в продукт. Вследствие этого Vmax уменьшается, т. е. в присутствии ингибитора пересечение прямой с осью ординат произойдет в более высокой точке (рис. 4.9). В той же мере возрастает и тангенс угла наклона прямой к оси абсцисс, равный КМ/VmaxI. КМ в отличие от Vmax не изменяется, поэтому неконкурентное ингибирование не может быть устранено увеличением концентрации субстрата.
Рис. 4.9. Неконкурентное ингибирование:
а – схема; б – графическое выражение по Лайнуиверу – Бэрку
Максимальная скорость реакции VmaxI в присутствии неконкурентного ингибитора описывается уравнением
 (4.7)
(4.7)
В частном случае бесконкурентного ингибирования, когда ингибитор связывается только с ES-комплексом и не связывается со свободным ферментом, на графике зависимости 1/V = f (1/[S]) прямые параллельны друг другу и пересекают оси ординат и абсцисс в разных точках (рис. 4.10).
Рис. 4.10. Бесконкурентное ингибирование:
а – схема; б – графическое выражение по Лайнуиверу – Бэрку
Необратимые ингибиторы– это высокореакционноспособные соединения различной химической природы, которые могут взаимодействовать с функционально важными группами активного центра, образуя прочные ковалентные связи. Это приводит к безвозвратной потере активности фермента. В связи с этим теория Михаэлиса – Ментен, основанная на предположении, что присоединение ингибитора к ферменту носит обратимый характер, в данном случае неприменима.
Примером необратимого ингибирования служит взаимодействие ферментов с ионами тяжелых металлов, которые присоединяются к сульфгидрильным группам цистеиновных остатков фермента и образуют при этом меркаптиды – практически недиссоциирующие соединения, либо ковалентная модификация фермента под действием алкилирующих агентов.
4.3. Методы выделения, очистки и определения активности ферментов
Будучи выделенными из клетки без повреждения нативной структуры, ферменты сохраняют свою активность, что делает возможным их использование в бесклеточных реакциях. Методы выделения и очистки ферментов описаны в главе 3. Следует отметить, что большинство ферментов являются лабильными белками, поэтому при выделении и очистке ферментов необходимо соблюдать целый ряд условий, предотвращающих их денатурацию и инактивацию под действием различных факторов. Эти условия для разных ферментов различны.
Активность фермента определяют по изменению начальной скорости реакции, измеряемой по количеству превращенного субстрата или образовавшегося продукта за единицу времени. Для этого используют спектрофотометрические, флуориметрические, манометрические, электродные и поляриметрические методы.
Наиболее предпочтительны спектрофотометрические методы вследствие своей простоты и высокой чувствительности. Они основаны на том, что под влиянием специфического действия фермента (кофермента) на субстрат образуется стехиометрическое количество продукта реакции (или уменьшается количество субстрата), обладающего характерным спектром поглощения в видимой или УФ-области спектра. По спектральным изменениям хромофора в ходе ферментативной реакции можно определить количество превращенного субстрата или образовавшегося продукта согласно закону Бугера – Ламберта – Бера:
 (4.8)
(4.8)
где С – молярная концентрация субстрата (продукта) или кофермента, М; Е – поглощение или экстинкция раствора; e - коэффициент молярной экстинкции поглощающего свет вещества при длине волны l, М-1 · см-1; l – толщина кюветы, см.
В настоящее время используются спектрофотометры, позволяющие непрерывно регистрировать изменение экстинкции во времени при определенной длине волны. При этом результаты записываются в виде непрерывной кинетической кривой и в автоматическом режиме производится расчет величины начальной скорости реакции – тангенса угла наклона касательной к кривой в нулевой точке.
Методы измерения активности ферментов делят на две группы – периодические и непрерывные. Последние более предпочтительны благодаря их быстроте и легкости выполнения, однако для многих ферментов нет таких способов измерений. Периодические методы можно использовать почти для всех ферментов, но они менее удобны.
При периодическом методе определения активности фермент инкубируют с субстратом в течение определенного периода времени, затем реакцию останавливают каким-либо способом, который ведет к мгновенной денатурации фермента, и измеряют количество образовавшегося продукта (или израсходованного субстрата). По значениям экстинкции, полученным при разных временах инкубации, строят кинетические кривые.
Возможны два варианта периодического метода: 1) из реакционной среды периодически отбирают пробы в строго фиксированные интервалы времени; 2) готовят серию параллельных проб, останавливая реакцию в них через различные интервалы времени.
Непрерывные методы измерения активности ферментов позволяют следить за ходом реакции во времени, не прерывая ее и регистрируя прирост продукта или убыль субстрата непрерывно в виде кинетических кривых.
В тех случаях, когда субстрат, продукт или кофермент не имеют характерных спектральных изменений в ходе ферментативной реакции, используют сопряженные методы − методы определения ферментативной активности в системе сопряженных реакций с применением вспомогательных ферментов, которые катализируют превращения продуктов первой ферментативной реакции с образованием соединений, имеющих хромофорные группы.
Условия проведения ферментативной реакции.
При проведении кинетических исследований необходимо знать:
1) общую стехиометрию катализируемой реакции;
2) возможную потребность в кофакторах;
3) зависимость активности фермента от концентраций субстрата и кофермента;
4) значение рН, соответствующее максимальной активности фермента;
5) область температур, при которых фермент устойчив и сохраняет высокую активность.
Как указывалось выше, скорость ферментативной реакции зависит от многих факторов. Это обстоятельство необходимо учитывать при выборе условий измерения активности фермента. Следует подобрать условия инкубации, при которых обеспечивается постоянство всех параметров, кроме измеряемого. Концентрация субстрата (и кофермента) должна превышать концентрацию насыщения (начальная скорость соответствует 0-му порядку реакции в отношении субстрата), с тем чтобы фактором, лимитирующим скорость реакции, была концентрация фермента. Начинать реакцию следует внесением фермента или одного из субстратов в минимальном объеме при интенсивном перемешивании реакционной смеси.
Обычно измерение скорости образования продукта реакции может быть проведено с большей точностью, чем измерение скорости исчезновения субстрата, так как для поддержания кинетики 0-го порядка субстрат должен присутствовать в высокой концентрации.
Для контроля степени очистки фермента его активность относят на 1 мг общего белка (удельная активность).
Определение активности алкогольдегидрогеназы (АДГ). Алкогольдегидрогеназа (алкоголь:НАД-оксидоредуктаза, КФ 1.1.1.1) катализирует конечную реакцию спиртового брожения – обратимое восстановление уксусного альдегида в этиловый спирт:
СН3СНО + НАДН · Н+ ⇄ СН3СН2ОН + НАД+
Фермент относится к классу оксидоредуктаз и обнаружен во многих тканях животных и растений, а также у микроорганизмов. АДГ пекарских дрожжей энергично окисляет первичные спирты с неразветвленной цепью и со значительно меньшей скоростью – спирты с разветвленной цепью, вторичные спирты, некоторые оксикислоты, аминоспирты и полиспирты. Молекула фермента (молекулярная масса ~150 кДа) состоит их четырех субъединиц, имеет четыре активных центра, содержит четыре атома прочно связанного цинка. АДГ является сульфгидрильным ферментом. В трис- и пирофосфатном буферных растворах оптимум рН равен 8,6.
Значения КМ:
· для НАД+ – 1,7 · 10-4 М; НАДН · Н+ – 2,3 · 10-5 М;
· этанола – 1,6 · 10-2 М; уксусного альдегида – 3,0 · 10-4 М.
Е2801% = 12,6.
Активность дегидрогеназ, нуждающихся в присутствии НАДН · Н+ и НАДФН · Н+ в качестве коферментов, измеряют с использованием непрерывных спектрофотометрических методов.
Активность АДГ определяют в реакции окисления этанола под действием фермента непосредственно по восстановлению НАД+, что регистрируют спектрофотометрически по увеличению экстинкции реакционной смеси при 340 нм. Коэффициент молярной экстинкции НАДН · Н+ при 340 нм равен 6,22 · 103 М-1 · см-1. При расчетах исходят из того, что при окислении−восстановлении 1 мкмоля кофермента при 340 нм в кювете толщиной 1 см в 1 мл реакционной смеси экстинкция меняется на 6,22 единиц.