Генные болезни аутосомно-рецессивного типа наследования
Фенилкетонурия (ФКУ)
Фенилкетонурия – наследственное заболевание обмена, характеризующееся поражением ЦНС и прогрессирующим, особенно в первые 2–3 года жизни, слабоумием. Фенотипически здоровые родители больного ребенка являются гетерозиготными носителями мутантного гена. Частота заболевания в Европе в среднем составляет 1:10 000 новорожденных, распространенность носителей гена в популяции 1:50. ФКУ наблюдается примерно у 1% умственно отсталых лиц.
Заболевание обусловлено мутацией гена, контролирующего синтез фермента фенилаланингидроксилазы, который обеспечивает превращение поступающего в организм с пищей фенилаланина в тирозин (рис. 21).
Нарушение последнего процесса приводит к резкому повышению содержания фенилаланина в сыворотке крови и спинномозговой жидкости, при этом отмечают дефицит тирозина, что определяет недостаточный синтез катехоламинов, гормона щитовидной железы и меланина, при недостаточном количестве которого наблюдается слабая пигментация кожи и волос. При ФКУ нарушается также обмен триптофана и синтез серотонина, что губительно действует на нормальное функционирование нервной системы. Ген РАН локализован на хромосоме 12q22.
Дети с ФКУ рождаются с полноценным головным мозгом, так как биохимические процессы плода осуществляются за счет процессов в организме матери. Возникающие после рождения биохимические нарушения оказывают токсическое воздействие на нервную систему, в результате чего нарушаются процессы миелинизации, развитие и рост мозга.
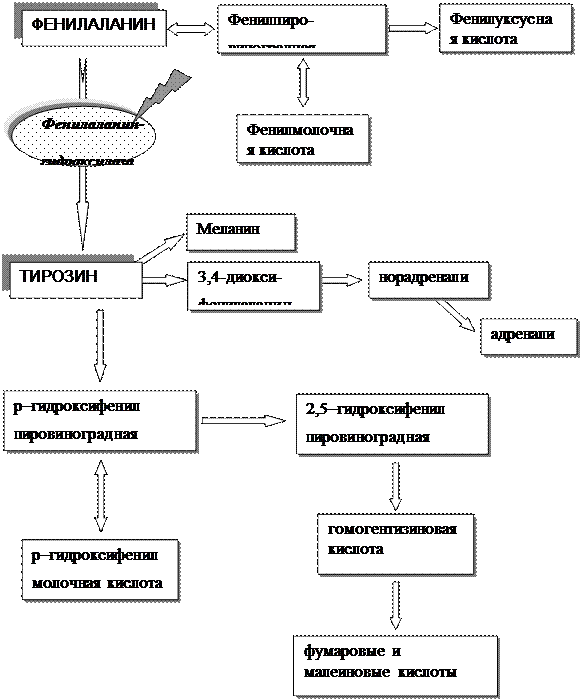
|
| Рис.21. Схема обмена фенилаланина и тирозина при фенилкетонурии |
Нарастание интеллектуального дефекта сочетается с отставанием в физическом развитии, часто с признаками умеренной микроцефалии. Характерен внешний вид больных (блондины со светлой кожей и голубыми глазами) и отдельные диспластические признаки (высокое небо, эпикант, деформация ушных раковин).
При этом отмечают следующие неврологические нарушения: мышечную гипертонию, повышение сухожильных рефлексов, гиперкинезы, тремор пальцев рук, атаксию, нарушения черепно–мозговой иннервации. В более редких случаях имеет место мышечная гипотония; судорожный синдром наблюдается у 20–50% больных.
Уровень интеллектуального развития колеблется от нормы до глубокой идиотии. Прогредиентность динамики слабоумия наиболее выражена в первые 2–3 года жизни. Больные отличаются инертностью, недостаточной целенаправленностью с характерными нарушениями внимания, памяти, недоразвитием гностических функций и пространственных представлений.
Отмечается также выраженное недоразвитие речи и нарушения звукопроизношения. Нарушения речи обычно сопоставимы с глубиной интеллектуального дефекта.
Гомоцистинурия
Гомоцистинурияобусловлена отсутствием или снижением активности фермента цистатионинсинтетазы, необходимого для синтеза цистатионина из гомоцистеина и серина (рис.22). Ген локализован на хромосоме 21q22.
Дефект генетически гетерогенен. Существуют две формы, различающиеся по отношению к витамину В6: пиридоксинзависимая и пиридоксинрезистентная. Описаны случаи заболевания, вызванные дефицитом других ферментов. Частота заболевания среди новорожденных колеблется от 1:80 000 до 1:180 000. Среди умственно-отсталых частота гомоцистинурии достигает 0.3%. В контингенте умственно отсталых с дефектами зрения – 2.6%.
 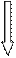                
| ||
| Рис.22. Обмен метионина при гомоцистинурии |
Клиническая картина полиморфна, но вместе с тем наиболее типичным симптомокомплексом считается сочетание умственной отсталости с дефектами зрения (эктопия хрусталика, катаракта, миопия) и костной системы (удлинение трубчатых костей при укороченном туловище, деформация суставов, деформация стоп, крыловидные лопатки). Внешними, наиболее выраженными признаками являются мягкие светлые волосы, голубые радужки, диспропорциональность телосложения с укорочением туловища и удлинением конечностей в сочетании со многими стигмами дисэмбриогенеза (воронкообразная грудная клетка, остеопороз костей и др.). Поражение соединительной ткани, механизм которого еще не ясен, определяет сходство гомоцистинурии с болезнью Марфана. Существует предположение о патогенетической роли в патогенезе гомоцистинурии дефицита меди.
Нервно–психические нарушения при этом заболевании отмечаются в 75% случаев. Описаны легкие (пограничные) и глубокие формы умственной отсталости с инертностью нервных процессов, недостаточной критичностью, расстройством речи. В ряде случаев отмечены двигательные нарушения в виде параличей и парезов. Нарушения речи включают общее недоразвитие, косноязычие, дизартрию.
Биохимическая диагностика направлена на качественное определение цистина и гомоцистина в моче, а также количественное определение метионина и гомоцистина в плазме на аминокислотном анализаторе. С целью ПД определяется активность цистатионинсинтетазы в культуре амниотических клеток. Лечение заключается в диете, бедной метионином. При пиридоксинзависимой форме заболевания эффективна терапия большими дозами витамина В6.
Галактоземия
Галактоземия обусловлена нарушением обмена галактозы. Ген, контролирующий синтез фермента галактозо-1-фосфат-уридилтрансфераза локализован на хромосоме 9р13. Генная мутация в гене приводит к дефициту фермента и к биохимическому блоку на этапе галактозо-1-фосфат. Биохимический катагенез болезни включает накопление галактозы и галактозо–1–фосфата в разных тканях и в крови. Вторичным эффектом является нарушение использования глюкозы в печени, почках и головном мозге (рис.23).
В выраженных случаях клинические проявления отмечаются уже с первых дней жизни ребенка в виде расстройств пищеварения и признаков интоксикации (гипотрофия, рвота, понос, отказ от кормления), желтухи с увеличением размеров печени, двусторонней врожденной катаракты. Иногда катаракта возникает несколько позже – на 4–7-й неделе жизни.
При некоторых моносимптомных формах эти проявления выражены нерезко, отмечаются либо умственная отсталость, либо катаракта в сочетании с непереносимостью молока. В более тяжелых случаях наблюдается сложный дефект – сочетание умственной отсталости с нарушением зрения (слепота). При рано начатом лечении диетой дети могут развиваться нормально.
Диагностируют галактоземию с использованием комплекса диагностических средств (в настоящее время создана система ее раннего выявления). Для предупреждения тяжелых нервно–психических отклонений разработана безлактозная диета.
          
      
| ||
| Рис.23. Генетические дефекты ферментов, катализирующих превращение галактозы в глюкозу |
Синдром Ушера
Распространенность синдрома Ушера среди детей с глубокой глухотой составляет от 3 до 10%. По данным европейского семинара по синдрому Ушера (1997 г.) люди с этим заболевание составляют до 6% всех глухих с рождения и до 50% всех слепоглухих взрослых.
Потеря зрения выявляется обычно в возрасте около 10 лет. Нарушение зрения медленно прогрессирует. Полная слепота может наступить в 50–60 лет. Офтальмологическое обследование обнаруживает типичный медленно прогрессирующий пигментный ретинит. Пигментный ретинит начинается скоплением гранул пигмента на глазном дне, распространяющихся по направлению к периферии. Поля зрения медленно сужаются и параллельно снижается острота зрения. К другим глазным симптомам относятся катаракта, глаукома.
Выявляется врожденная нейросенсорная потеря слуха от умеренной до резко выраженной степени. У больных отмечается атрофия кортиева органа и эпителия внутреннего и наружного желобка в нижней части базального завитка улитки. Дегенеративные изменения в верхнем завитке. Имеется резкая атрофия спирального узла, его периферических и центральных волокон.
Обнаруживаются дефекты вестибулярной системы, которые выражаются в нарушении равновесия при ходьбе. Нарушение равновесия возможно обусловлено нарушениями лабиринта, а не мозжечковой патологией. У больных, кроме основных симптомов, выявляются также психозы, агрессивность, периодические депрессии, у 25% - умственная отсталость.
Синдром наследуется по аутосомно-рецессивному типу. Ген локализован на хромосоме 14q.
Сочетание глухоты с пигментным ретинитом впервые было описано А. Графе в 1858 г., а генетическую природу этого синдрома установил С. Ушер в 1914 г. Выявлено, что один из 100 человек является носителем гена синдрома Ушера. У гетерозигот могут быть выявлены отсутствие реакции на вращение, повышение порога темновой адаптации или незначительное снижение зрения.
Своевременное выявление у больных пигментного ретинита и создание адекватных педагогических условий предотвращают стрессовые состояния, связанные у глухого человека с потерей зрения. Методы лечения отсутствуют.