Диагностика синдрома Марфана
Синдром Марфана
| Синдромом Марфана называется аутосомно-доминантная генетическая болезнь, которая поражает соединительную ткань и которая связана с нарушениями функционирования соединительной ткани. Данный синдром характеризуется наличием у пациентов непропорционально длинных конечностей. Синдром Марфана - дифференцированная форма врожденной соединительнотканной недостаточности, характеризующаяся разнообразными проявлениями скелетной, сердечно-сосудистой и глазной патологии. У больных с синдромом Марфана отмечаются гигантизм, долихостеномелия и арахнодактилия, аневризмы аорты, миопия, эктопия хрусталика, деформация грудины, кифосколиоз, плоскостопие, протрузия вертлужной впадины, эктазия твердой мозговой оболочки. Диагноз синдрома Марфана основан на семейном анамнезе, результатах функционального, офтальмологического, рентгенологического и генетического исследований. Лечение при синдроме Марфана включает консервативную и хирургическую коррекцию сердечно-сосудистых нарушений, поражений скелета и органа зрения. подробнее |
У таких больных длинные худые пальцы, худое телосложение. Кроме того, у них обычно имеются сердечно сосудистые пороки, которые зачастую проявляются в виде пороков аорты и клапанов. Самыми серьезными последствиями болезни являются повреждения клапанов сердца, а так же нарушение стенки аорты. Кроме того, синдром Марфана может влиять на глаза, легкие, скелет, твердую оболочку спинного мозга и твердое небо. Поэтому данному заболеванию характерен полиморфизм клинических проявлений.
Синдром Марфана может проявляться как в умеренной, так и в весьма тяжелой форме. Люди с данным заболеванием обычно имеют высокий рост и длинные худые конечности.
Свое название заболевание получило от имени французского педиатра Антуана Марфана. Именно он в 1896 году впервые описал симптомы этого заболевания, когда изучил характерные черты синдрома у маленькой девочки. Ген, который вызывает заболевание, был открыт не сразу, а только в 1991 году Франческо Рамиресом, работающем в Нью-Йорке.
Причины
Синдром Марфана встречается примерно у 1 человека из 5 тысяч человек. Поэтому он считается довольно редким заболеванием. Согласно проведенным исследованиям данное заболевание вызывается мутацией определенного гена, а именно гена белка фибриллина, который находится в пятнадцатой хромосоме. Эта мутация гена приводит к появлению нарушениям в нормальной выработке фибриллина и аномалиям в его структуре. Чаще всего ген данного синдрома передается детям от их родителей, которые так же имеют данное заболевание. Если у одного из родителей есть синдром Марфана, то в 50% случаев ребенок так же родится с этим заболеванием.
Симптомы синдрома Марфана
Каких-то уникальных симптомов, которые характерны этому заболеванию, не существует. Для постановки диагноза следует учитывать наличие у больного сочетания нескольких признаков. К этим признакам относятся: длинные конечности, аневризма корня аорты и дислокация хрусталика глаза. Всего насчитывается более 30 клинических признаков данной патологии. Все эти признаки в основном характеризуются изменениями скелета, суставов и кожи. Считается, что высока вероятность передачи синдрома по наследству.
Основная часть симптомов синдрома Марфана связана с костной системой. Поэтому не удивительно, что большинство людей с этим синдром достаточно высокого роста. Часть из них имеет длинные конечности и длинные тонкие пальцы рук и ног. Отношение размаха рук к росту обычно составляет 1,05. Кроме непропорциональных конечностей и высокого роста у больных могут возникать и другие нарушения в костной системе. Самым распространенным нарушением является искривление позвоночника. Кроме того, возможна воронкообразная либо килевидная деформация грудной клетки, чрезмерная гибкость суставов, неправильный прикус, высокое небо, плоскостопие, сутулость. У некоторых больных суставы пальцев на ногах согнуты, чем напоминают молоток.
Так же возможно появление беспричинных растяжек на коже. Часть пациентов отмечает боли в суставе, боли в мышцах и костях. Кроме того, у таких больных возможно нарушение и расстройство речи, что вызывается малым размером челюсти и высоким небом. Помимо этого, существует вероятность развития в раннем возрасте остеоартрита.
К малым критериям нарушений в костной системе относится неполное разгибание локтевых суставов, плоскостопие, симптом запястья и симптом большого пальца, протрузия вертлужной впадины, сколиоз, разболтанность суставов и «птичье лицо». О присутствии синдрома можно говорить, если у больного присутствует хотя бы два больших критерия заболевания или одно проявление большого и два проявления малых критерия.
Синдром Марфана так же может отрицательно влиять на зрение и глаза. У таких пациентов чаще всего диагностируется астигматизм и близорукость. В некоторых случаях у таких людей может присутствовать наоборот дальнозоркость. Кроме того, в 80% случаев отмечается неправильное положение хрусталика в одном или обоих глазах. Эти нарушения легко может выявить врач офтальмолог с помощью щелевой лампы. В некоторых случаях проблемы со зрением появляются только после ослабления соединительной ткани, которое происходит из-за расслоения сетчатки. У части больных в раннем возрасте наблюдается такая офтальмологическая проблема, как ранняя глаукома. Большим критерием наличия изменений в глазах при данном синдроме является присутствие подвывиха хрусталика. При этом он смещен вверх и имеет дефект цинковой связи. К малым критериям относят уплощение роговицы, гипоплазию радужной оболочки, гипоплазию ресничной мышцы, а так же увеличение размера глазного яблока. Если есть в наличии хотя бы два критерия, то уже можно говорить о вовлечении глаз в синдром.
Самыми серьезными признаками и симптомами данного заболевания считаются нарушения нормальной деятельности сердечно сосудистой системы. Признаками таких нарушений могут быть такие симптомы, как отдышка, повышенная утомляемость, нарушение ритма сердца, учащенное сердцебиение и стенокардия. Нередко у больных синдромом, из-за таких нарушений, руки и ноги практически всегда холодные. Если при диагностике выявляются шумы в сердце и изменения в ЭКГ или присутствуют симптомы стенокардии, то пациента обычно отправляют на дальнейшее обследование. Причиной возникновения регургитации может стать кистозная медиальная дегенерация клапанов, возникающая из-за пролапса митрального и аортального клапана.
Большим критериями диагностики заболевания считается расширение восходящей аорты и расслаивающая аневризма восходящей аорты. К малым критериям относится пролапс митрального клапана, расширение легочного ствола и кальциноз митрального кольца. Последние два учитываются только до 40 лет. Так же к малым критериям относят расширение или расслаивающаяся аневризма брюшной и грудной аорты. Этот критерий учитывается до 50 лет. Если имеются любые из малых критериев, то можно говорить о вовлечении сердечнососудистой системы в синдром Марфана.
Стоит почеркнуть, что ведущими критериями при диагностике болезни, считаются как расширение аорта и аневризма аорта. Тем не менее, в некоторых случаях у больного не выявляется каких-либо проблем с сердцем. Несмотря на это, ослабление соединительной ткани, вызывает аневризму или расслоение восходящей части аорты. Эта патология требует оперативного лечения. При расслоении аорты больной жалуется на боли в спине или груди. Болевые ощущения согут приводить к тому, что у больного будет возникать ощущение надрыва. Из-за нарушения функциональности соединительной ткани искусственные митральные клапана могут попросту не приживаться в организме больного. По этой причине при синдроме Марфана нужно быть внимательным при лечении клапанов сердца. Лучше всего применять те способы лечения, которые направлены не на замену клапана, а на восстановление нормальной функциональности клапана.
Стоит выделить случаи беременности у женщин с синдромом Марфана. Если у женщины нет никаких видимых нарушений в работе сердечнососудистой системы, то риск расслоения аорты все равно достаточно высокий. Это может привести к летальному исходу, даже если будет проведено своевременное лечение. По этой причине, женщинам с синдромом Марфана, перед тем как забеременеть, необходимо пройти тщательное обследование и получить заключение врача. Если врач разрешил беременеть, то для определения диаметра аорты необходимо каждые 6-10 недель во время беременности выполнять эхокардиографию. Если соблюдать все рекомендации врача, то роды обычно проходят без особых осложнений естественным путем.
Люди с синдромом Марфана находятся в группе риска возникновения скопления воздуха в плевральной полости (пневмоторакс). В этом случае воздух выходит из легких и занимает привальную полость между легкими и грудной клеткой. Это приводит к тому, что легкие сжимаются. Больной со скоплением воздуха в плевральной полости обычно жалуется на резкую боль в груди. При этом он дышит преимущественно поверхностно и часто. У больного наблюдается выраженная отдышка. Нередко у таких людей отмечается синюшность или бледность кожных покровов, особенно лица. В случае отсутствия адекватного лечения это может привести к летальному исходу. Синдром Марфана так же может быть связан с апноэ во сне. При этом у человека на время прекращается вентиляция легких более чем на десять секунд. Как таковых больших диагностических критериев наличия болезни в случае нарушений в легких нет. Среди малых критериев нужно отметить буллы в верхушке легкого и спонтанный пневмоторакс.
Синдром Марфана может негативно сказаться на качестве жизни человека, если возникнет дуральная ектазия, которая представляет собой ослабление и растяжение соединительной ткани дурального мешка мембраны, окутывающей спинной мозг. Вообще среди поражений нервной системы большими диагностическими критериями синдрома обладает эктазия твердой мозговой оболочки, возникающая в пояснично-крестцовом отделе. Эти нарушения могут быть обнаружены с помощью МРТ (магнитно-резонансной) или КТ (компьютерной томографии).
· Симптомы синдрома Марфана
Синдром Марфана
· Причины синдрома Марфана
· Классификация синдрома Марфана
· Прогноз и профилактика синдрома Марфана
· Синдром Марфана - лечение в Москве
 Синдром Марфана - системное недоразвитие соединительной ткани в эмбриональном и постнатальном периодах, обусловленное структурными дефектами коллагена и сопровождающееся преимущественным поражением опорно-двигательного аппарата, глаз, сердечно-сосудистой системы. Синдром Марфана - одна из наиболее распространенных наследственных коллагенопатий синдромального характера. Частота встречаемости синдрома Марфана в популяции невысока: по данным различных авторов составляет 1 случай на 10000-20000 человек, без расовой и половой детерминированности.1
Синдром Марфана - системное недоразвитие соединительной ткани в эмбриональном и постнатальном периодах, обусловленное структурными дефектами коллагена и сопровождающееся преимущественным поражением опорно-двигательного аппарата, глаз, сердечно-сосудистой системы. Синдром Марфана - одна из наиболее распространенных наследственных коллагенопатий синдромального характера. Частота встречаемости синдрома Марфана в популяции невысока: по данным различных авторов составляет 1 случай на 10000-20000 человек, без расовой и половой детерминированности.1
Синдром Марфана относится к моногенным болезням соединительной ткани - группе различных по происхождению нозологических форм, которые объединяют наследственные нарушения обмена соединительной ткани. Большая часть этой патологии обусловлена нарушением ферментных систем, контролирующих синтез структурных белков из которых происходит синтез соединительной ткани. Почти все эти болезни, входящие в состав синдрома, приводят к тяжелым инвалидизирующим расстройствам.
Синдром Марфана впервые описан Вильямсом в 1876 г. Свое название заболевание получило от французского педиатра Марфана, наблюдавшего девочку с характерным симптомокомплексом болезни 20 лет спустя.
____________________________________________________
1 http://www.krasotaimedicina.ru/diseases/children/marfan-syndrome
Существует интересный факт, что первая девушка модель - Лесли Хорнби, которая послужила прототипом образа всех моделей, имела синдром Марфана. Так, установлено, что ряд всемирно известных людей страдали синдромом Марфана, среди них следует упомянуть президента США А. Линкольна и великого скрипача Паганини.
 |
Лесли Хорнби (Твигги)
Симптомы синдрома Марфана
Клиническая картина заболевания характеризуется поражением многих жизненно важных органов и систем: опорно-двигательного аппарата, сердечно-сосудистой системы, органов дыхания и зрения, ЦНС.
Так, среди конституциональных особенностей и нарушений скелета наиболее часто встречаются долихопластический (астенический) тип, высокий рост (как правило, выше 180см) при выраженном дефиците массы тела (обычно ниже 50 кг.), арахнодактилия («паучьи» пальцы) кистей и стоп, кифосколиоз, воронкообразная или килевидная деформации грудной клетки, плоскостопие, узкий лицевой скелет, «готическое» небо. Не обязательно у одного человека встречаются все эти признаки, это лишь наиболее распространееные

Арахнодактилия
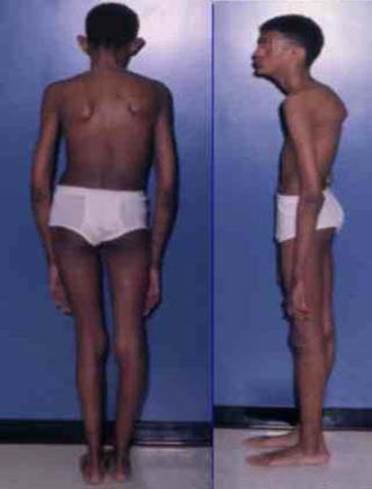
Внешний вид больного с синдромом Марфана
Типичны также антимонголоидный разрез глаз, «крупный» нос, большие низкорасположенные ушные раковины, «птичье» выражение лица. У новорожденных из перечисленных особенностей скелета, как правило, выявляются только долихопластический тип и арахнодактилия. Остальные симптомы формируются в более поздние периоды развития (обычно в течение первых семи лет жизни ребенка).
Поражение сердца и сосудов - один из кардинальных признаков синдрома Марфана. Наиболее типичны среди них - пролапс митрального клапана и аневризма аорты. Сердечно-сосудистые нарушения регистрируются уже на первом-втором годах жизни ребенка, при этом отмечается постепенное увеличение диаметра аорты, достигающее критических размеров (до 6 см и более), чаще в возрасте от 16 до 45 лет.
Грозным осложнением аневризмы аорты является расслоение ее стенок, которое может быстро прогрессировать, захватывая всю длину аорты и отходящие от нее сосуды. Такие осложнения, как правило, заканчиваются летально.
Бронхолегочная система также вовлекается в патологический процесс при синдроме Марфана. Предпосылкой для этого являются механическое сдавление дыхательных путей при деформациях грудной клетки и изменения соединительнотканных структур легочной ткани. Нарушения органов дыхания в виде спонтанного пневмоторакса, легочной эмфиземы, инфаркта легкого встречаются с частотой от 10 до 75%. Наряду с этим, имеются сведения о врожденном недоразвитии одной из долей легкого, поликистозе легких, врожденной буллезной эмфиземе, двусторонних бронхоэктазах.
К наиболее типичной патологии органа зрения при синдроме Марфана относится вывих и подвывих хрусталика (вследствие слабости цинновой связки). Как правило, эта патология сочетается с миопией или гиперметропией высокой степени. Подвывих хрусталиков диагностируется обычно на 1-5 годах жизни, а иногда даже в 7 лет при оформлении ребенка в школу. Реже встречаются вторичная глаукома, катаракта, отслойка сетчатки. Эти изменения чаще выявляются у больных более старшего возраста 15- 40 лет.
IQ (коэффициент интеллектуального развития) у большинства детей с синдромом Марфана обычно соответствует норме – 85-115 единиц. Встречаются лица с очень высоким интеллектом, у которых IQ превышает верхнюю границу нормы -115 ед. Однако может иметь место определенное своеобразие психических процессов, которое проявляется в неравномерной интеллектуальной деятельности, а также в личностных особенностях больных (раздражительности, плаксивости, завышенной самооценке).
Для всех детей с синдромом Марфана характерна низкая переносимость физической нагрузки, которая нередко сопровождается болями в мышцах. Возможны также периодические приступы мигренеподобной головной боли, возникающей, как правило, на фоне или после эмоционально-физических нагрузок. Эти признаки заболевания в сочетании со слабостью, гипотонией и гипоплазией мышечной ткани, а также нарушениями показателей физического развития служат свидетельством изменения функции митохондрий (нарушением процессов клеточной биоэнергетики).
Причины синдрома Марфана
Синдром Марфана относится к врожденным аномалиям, наследуемым по аутосомно-доминантному типу, с выраженным плейотропизмом, варьирующей экспрессивностью и высокой пенетрантностью. В основе синдрома Марфана лежат мутации в гене FBN1, отвечающем за синтез фибриллина – важнейшего структурного белка межклеточного матрикса, придающего эластичность и сократимость соединительной ткани. Аномалия и дефицит фибриллина при синдроме Марфана приводят к нарушению формирования волокнистых структур, потере прочности и упругости соединительной ткани, невозможности выдерживать физиологические нагрузки. Гистологическим изменениям в большей степени подвержены стенки сосудов эластического типа и связочный аппарат (в первую очередь, аорта и цинновая связка глаза, содержащие наибольшее количество фибриллина).
Широкий фенотипический спектр синдрома Марфана (от легких форм, трудно отличимых от нормы до тяжелых, быстропрогрессирующих) объясняется разнообразием мутаций в гене FBN1 (более 1000 видов), а также присутствием мутаций в других генах (например, в гене трансформирующего фактора роста - TGFBR-2). При генетическом исследовании в 75% случаев синдрома Марфана выявляется семейный тип наследования, в остальных - первичная мутация. Риск рождения ребенка с синдромом Марфана возрастает с увеличением возраста отца (особенно после 35 лет).3
Диагностика синдрома Марфана
Диагностика синдрома Марфана базируется на генеалогических данных (составление и анализ родословных) и анализе морфофенотипа, который включает изучение физического, нервно-психического развития детей и состояния физического развития больных проводится с помощью перцентильных шкал Стюарта.
О пропорциональности или гармоничности отдельных частей тела судят путем использования индекса Дю Ранта-Лайнера, который вычисляется по формуле А/В х 100, где А - отношение фактической массы тела к 50 перцентилю массы, соответствующей росту больного, а В - отношение фактической длины тела к 50 перцентилю роста соответствующего возраста. Индекс количественно отражает вариации физического развития. При этом показатель 89 и ниже соответствует высокому росту при дефиците массы тела, показатели - 110-119 - избыточной массе тела, свыше 120 – ожирению. Для детей, свыше 120 – ожирению. Для детей с синдромом Марфана индекс Дю Ранта-Лайнера, как правило, составляет 51-81.
По резолюции совещания, посвященного синдрому Марфана, для постановки диагноза необходимо наличие минимум одного из пяти основных симптомов заболевания (вывих хрусталиков, аневризма аорты, арахнодактилия, деформация грудины, кифосколиоз) и двух дополнительных (миопия, пролапс митрального клапана, умеренная гиперподвижность суставов, высокий рост, плоскостопие, стрии, пневмоторакс).
Установлено что в 90% всех случаев синдрома Марфана трудностей в постановке правильного диагноза, как правило, не возникает. Однако в 10% - диагностика затруднена. В подобных ситуациях особенно необходимо чрезвычайно тщательное обследование максимально большего числа родственников больного. Программа обследования для таких семей должна обязательно включать консультативные, осмотры окулиста, кардиолога и проведение эхокардиографии.
В диагностике синдрома Марфана широко используются также результаты рентгено-функциональных методов исследования. Так, для оценки арахнодактилии используют показатели метакарпального индекса (отношение длины к ширине второй-пятой метакарпальных костей), вычисляемого по рентгенограмме правой кисти. У больных с синдромом Марфана наблюдается увеличение этого показателя до 8,0-11,0 при норме 6,4-7,9.
Характер и степень тяжести сердечно сосудистой патологии оценивают по данным эхокардиографии, ЭКГ, холтеровскому мониторированию. Анализ состояния бронхолегочной системы проводят по результатам исследования функции внешнего дыхания. У подавляющего числа детей с синдромом Марфана регистрируются изменения этих показателей проявляющиеся в нарушении механики дыхания, вздутии легочной ткани, неравномерности распределения в легких вдыхаемого воздуха гиперкапнии. У детей с синдромом Марфана обнаруживается снижение репарационной способности ДНК-лимфоцитов, что необходимо принимать во внимание при рентгенологическом обследовании выборе профессии и места жительства больных.
_________________________________________________________
3 http://www.krasotaimedicina.ru/diseases/children/marfan-syndrome
При синдроме Марфана имеет место увеличение (в два раза и более) выведения с мочой гпикозаминогликанов и их фракций, при этом особенно резко возрастает почечная экскреция хондроитин-4-6-сульфатов и в меньшей степени - гиалоурановый кислоты и гепарин-сульфата.
Дифференциальную диагностику проводят со сходными синдромами и генетическими заболеваниями. В качестве критерий сравнения выступает ряд признаков проявления различных заболеваний.