Метод кислотно-основного титрування
ВСТУП
Харчові добавки – це природні, ідентичні природним або штучні (синтетичні) речовини, попередньо введені в харчову сировину, напівпродукти чи готові продукти з метою збільшення терміну їx зберігання або надання їм заданих властивостей.
Харчові добавки поділяють на декілька груп:
– речовини, що регулюють смак продукту (ароматизатори, смакові добавки, підсолоджувальні речовини, кислоти і регулятори кислотності);
– речовини, які покращують зовнішній вигляд продукту (барвники, стабілізатори кольору, відбілювачі);
– речовини, що регулюють консистенцію і формують текстуру (згущувачі, гелеутворювачі, стабілізатори, емульгатори тощо);
– речовини, що збільшують термін зберігання продуктів (консерванти, антиоксиданти).
Завдання дисципліни «Хімія і технологія харчових добавок» полягає в тому, щоб дати студентам міцні знання з таких питань, як класифікація основних типів харчових добавок, методи отримання харчових добавок шляхом синтезу або виділення з природних джерел, залежність фізико-хімічних і біологічних властивостей харчових добавок від структури їх молекул, вплив хімічної будови на біологічні властивості, основні шляхи практичного застосування харчових добавок, можливість одержання їх аналогів синтетичним або мікробіологічним методами. Крім того, необхідно навчити студентів самостійно виконувати експериментальні роботи з виділення, ідентифікації, аналізу та синтезу простих за будовою представників основних типів харчових добавок, досліджувати їх фізико-хімічні властивості.
Лабораторна робота 1
Синтез регуляторів рН – сполук кальцію
Мета роботи: вивчення промислових і лабораторних методів отримання підлужнювальних речовин: оксиду та карбонату кальцію.
Реактиви: СаСО3, Са(NO3)2, (NH4)2CO3, дифеніламін.
Оксид кальцію (Е 529)
Оксид кальцію (СаО) застосовується як регулятор кислотності, покращувач якості борошна та хліба.
Метод отримання:
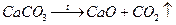 .
.
Методика роботи. Наважку СаСО3 (ч.д.а.) масою 1 г прокалюють при 970–1050 С протягом 2–3 год.
Кальцію карбонат (Е 170i)
Карбонат кальцію (СаСО3) застосовується в якості добавки, яка попереджує злежування, комкування, та як стабілізатор.
Метод отримання
Вуглекислий кальцій отримують дією вуглекислого амонію на азотнокислий кальцій:
Са(NO3)2 + (NH4)2CO3 = CaCO3↓ + 2NH4NO3.
Методика роботи. Окремо готують розчини 2,5 г Са(NO3)2·4Н2О (ч.д.а.) в 25 мл води та 1,18 г (NH4)2СО3 (ч.) в 25 мл води. Отримані розчини при перемішуванні вливають з однаковою швидкістю в склянку з 20 мл води. Після відстоювання перевіряють повноту осадження, додаючи при необхідності ще трохи (NH4)2CO3. Осад отриманого карбонату кальцію відфільтровують та промивають на фільтрі повільним потоком дистильованої води до повного видалення NO3- (проводять реакцію з дифеніламіном, поява синього забарвлення свідчить про наявність нітрат-іонів). Промитий осад карбонату кальцію сушать при 110 С протягом 2 год.
Контрольні запитання
1. Яку роль відіграють сполуки кальцію як харчові добавки?
2. Які властивості цих сполук?
3. Якими методами отримують оксид кальцію?
4. Якими методами отримують карбонат кальцію?
Лабораторна робота 2
Методи визначення підлужнювачів – сполук кальцію
Мета роботи: провести якісний і кількісний аналіз отриманих сполук кальцію.
Реактиви: СаСО3, НСlрозв., NH4OH, (NH4)2C2O4, NaOH, трилон Б, індикатори мурексид і метиловий жовтогарячий.
Реакції на автентичність
1. Визначення кальцію
Са2+ + С2О42- ® СаС2О4¯.
Методика роботи. Наважку оксиду кальцію масою 0,1 г розчиняють у розведеній соляній кислоті. До отриманого розчину додають декілька крапель розчину аміаку до нейтрального середовища (перевіряють за допомогою універсального індикаторного папірця) і потім додають 1 мл розчину оксалату амонію. У присутності іонів кальцію утворюється білий осад, нерозчинний у розведеній ацетатній кислоті і розчині аміаку, розчинний у розведених мінеральних кислотах.
2. Визначення карбонатів
СО32- + 2Н+ ® СО2 + Н2О;
Са(ОН)2 + СО2 ® СаСО3¯ + Н2О.
Методика роботи. До 0,01 г карбонату кальцію додають 0,5 мл розведеної соляної кислоти; виділяється вуглекислий газ, який утворює білий осад при пропусканні через вапняну воду.
Кількісне визначення сполук кальцію
1. Метод комплексонометрії
Кальцій стехіометрично реагує з трилоном Б з утворенням стійкої внутрішньокомплексної сполуки. Титрування проводять у лужному середовищі з індикатором мурексидом.
Сa2+ + [H2Y]2- → [CaY]2- + 2H+;
Сa2+ + Ind- → CaInd+;
CaInd+ + [H2Y]2- → [CaY]2- + 2H+ + Ind-.
Методика роботи. Наважку малорозчинного у воді CaO або CaCO3 (0,05–0,1 г) переносять у конічну колбу, додають 20–30 мл 0,1 н. розчину HCl та нагрівають на повільному вогні до повного розчинення. До отриманого розчину додають 10 мл 1 н. розчину NaOH. Розводять дистильованою водою до 100 мл та після ретельного перемішування додають індикатор – мурексид. Потім при ретельному перемішуванні повільно титрують отриманий розчин 0,1 н. розчином трилону Б до переходу забарвлення із червоного у фіолетове.
Розраховують вміст CaO або CaCO3 за формулою:
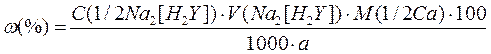 ,
,
де М (1/2Са) – еквівалентна маса СаO або СаCO3;
a – маса наважки, г;
VNa2[H2Y] – об’єм розчину трилону, витрачений на титрування, мл.
2. Метод кислотно-основного титрування
СаO взаємодіє з водою з утворенням Ca(OH)2, а CaCO3 малорозчинний у воді, але добре розчиняється в сильних кислотах. Тому вміст кальцію в цих сполуках можна визначити кислотно-основним титруванням за методом залишків.
СаO + HClнадл. ® СаCl2 + H2O;
СаCO3 + HClнадл. ® СаCl2 + H2O + CO2↑;
HClзал. + NaOH ® NaCl + H2O.
Методика роботи. Наважку речовини 0,05 г переносять у конічну колбу та додають надлишок 0,1 н. розчину HCl (30 мл). Суміш нагрівають на водяній бані до повного розчинення, розчин охолоджують, додають 100 мл дистильованої води та 1–2 краплі індикатора метилового жовтогарячого. Надлишок кислоти титрують стандартним розчином 0,1 н. NaOH.
Вміст СаO або СаCO3 розраховують за формулою:

де VHCl – загальний об’єм розчину кислоти, мл;
VNaOH – об’єм розчину лугу, витрачений на титрування, мл;
М(1/2Са) – еквівалентна маса СаO або СаCO3;
a – маса наважки, г.
Контрольні запитання
1. Які реакції використовуються при визначенні автентичності підлужнювачів – сполук кальцію?
2. У чому полягає сутність комплексонометричного визначення сполук кальцію в харчових продуктах?
3. Наведіть реакції, які використовуються при кислотно-основному визначенні сполук кальцію?
Лабораторна робота 3
Синтез регуляторів рН – сполук магнію
Мета роботи: вивчення промислових і лабораторних методів отримання підлужнювальних речовин: оксиду та карбонату магнію.
Реактиви: MgCl2·6Н2О, Na2CO3·10Н2О, індикатор фенолфталеїн.
Оксид магнію (Е 530)
Оксид магнію (МgО) застосовується як добавка, що попереджує злежування та комкування.
Методи отримання
Метод базується на отриманні основного вуглекислого магнію, який потім прожарюють при високій температурі:
2MgCl2 + 2Na2CO3 + H2O = Mg2(OH)2CO3↓ + 4NaCl + CO2↑;
Mg2(OH)2CO3 = 2MgO + CO2↑ + H2O↑.
Методика роботи. Розчиняють ~ 2,5 г MgCl2·6Н2О в 50 мл води, вносять 0,015 г MgО перемішують, дають відстоятися та фільтрують. Фільтрат нагрівають до 80–85 С і приливають тонкою цівкою холодний відфільтрований розчин (4,2 г Na2CO3 10Н2О (ч.д.а.) в 50 мл води) до тих пір, поки реакція фільтрату стане слабколужною за фенолфталеїном (до блідо-рожевого забарвлення). Розчин нагрівають 10 хв та фільтрують. Осад промивають в 25 мл гарячої води та сушать протягом 1 год при 110-120ºС. Отриману сіль прожарюють в тиглі при 600-750ºС протягом 0,5-1 год.
Отримання магнію вуглекислого основного
Основний вуглекислий магній отримують при взаємодії вуглекислого натрію із сіллю магнію.
Методика роботи. Розчиняють 6 – 6,5 г MgCl2·6Н2О (техн.) в 20 мл води, додають 0,1 мл конценрованого розчину NH4OH та нагрівають до 60ºС. Суміші дають відстоятися та осад механічних домішок і Fe(OH)3 відфільтровують.
Окремо розчиняють 3 г Na2CO3 (техн.) в 17,5 мл гарячої води; додають 0,10-0,12 мл 40%-го розчину NaOH та кип’ятять. Суміші дають відстоятися та фільтрують. Отриманий фільтрат нагрівають до 60ºС та приливають тонкою цівкою до гарячого розчину MgCl2 до повного осадження основного вуглекислого магнію (відфільтрована проба рідини не повинна давати осаду з BaCl2 та з Na2CO3). Осад відфільтровують, промивають водою до видалення Cl- (допустима лише дуже невелика опалесценція проби з AgNO3) та сушать при 70 С, краще у вакуумі.
Контрольні запитання
1. Яку роль відіграють сполуки магнію як харчові добавки?
2. Які властивості цих сполук?
3. Якими методами отримують оксид магнію?
4. Якими методами отримують карбонат магнію?
Лабораторна робота 4
Методи визначення якості та кількісного вмісту
підлужнювачів – сполук магнію
Мета роботи: провести якісний і кількісний аналіз отриманих сполук магнію.
Реактиви: MgO, HCl, NH4OH, Na2НPO4, трилон Б, індикатор еріохром чорний Т та метиловий жовтогарячий, аміачний буфер, NaOH.
Якісне визначення сполук магнію
1. Визначення магнію
MgO + 2HCl → MgCl2 + H2O;
Mg2+ + NH4OH + НPO42- → MgNH4PO4¯ + H2O.
Методика роботи. 0,01 г оксиду магнію розчиняють у суміші з 0,5 мл розведеної хлоридної кислоти та 0,5 мл води. До отриманого розчину додають 1 мл розчину хлориду амонію, 0,5 мл розчину гідрофосфату натрію та 1 мл розчину аміаку. Утворюється білий кристалічний осад, розчинний в оцтовій кислоті.
2. Визначення карбонатів
СО32- + 2Н+ ® СО2 + Н2О;
Са(ОН)2 + СО2 ® СаСО3¯ + Н2О.
Методика роботи: До 0,01 г карбонату магнію додають 0,5 мл розведеної хлоридної кислоти; виділяється вуглекислий газ, який утворює білий осад при пропусканні через вапняну воду.
Кількісне визначення сполук магнію
Метод кислотно-основного титрування
МgO та MgCO3 добре розчиняються в сильних кислотах. Тому вміст магнію в цих сполуках можна визначити кислотно-основним титруванням за методом залишків.
MgO + HClнадл. ® MgCl2 + H2O;
MgCO3 + HClнадл. ® MgCl2 + H2O + CO2↑;
HClзал. + NaOH ® NaCl + H2O.
Методика роботи. Наважку речовини 0,05 г переносять у конічну колбу та додають надлишок 0,1 н. розчину HCl (до 20 мл). Суміш нагрівають на водяній бані до повного розчинення, розчин охолоджують, додають 1 краплю індикатора метилового жовтогарячого. Надлишок кислоти титрують стандартним розчином 0,1 н. NaOH до переходу жовтого кольору в рожевий.
Вміст МgO або MgCO3 розраховують за формулою:

де VHCl – загальний об’єм розчину кислоти, мл;
VNaOH – об’єм розчину лугу, витрачений на титрування, мл;
М(1/2Mg) – еквівалентна маса МgO або MgCO3;
a – маса наважки, г.
2. Метод комплексонометрії
Магній стехіометрично реагує з трилоном Б з утворенням стійкої внутрішньокомплексної сполуки.

Титрування проводять у лужному середовищі з індикатором еріохромом чорним Т.
Mg2+ + [H2Y]2- → [MgY]2- + 2H+;
Mg2+ + Ind- → MgInd+;
MgInd+ + [H2Y]2- → [MgY]2- + 2H+ + Ind-.
Методика роботи. Наважку малорозчинного у воді MgO або MgCO3 (0,05–0,1 г) переносять у конічну колбу, додають 20 мл 0,1 н. розчину HCl та нагрівають на повільному вогні до повного розчинення. Додають NH4OH (1:1) до слабкого запаху аміаку (15–20 крапель), додають 10 мл аміачного буферного розчину (рН = 9) та індикатор – еріохром чорний Т. При ретельному перемішуванні титрують з бюретки розчином трилону Б відомої концентрації до переходу забарвлення з червоного в синє.
Вміст МgO або MgCO3 розраховують за формулою:
 ,
,
де М(1/2Mg) – еквівалентна маса МgO або MgCO3;
a – маса наважки, г.
Контрольні запитання
1. Які реакції використовуються при визначенні автентичності підлужнювачів – сполук магнію?
2. У чому полягає сутність кислотно-основного визначення сполук магнію?
3. Наведіть реакції, які використовуються при комплексонометричному визначенні сполук магнію в продуктах харчування?
Лабораторна робота 5
Методи отримання й ідентифікації консервантів
Мета роботи: вивчення методів отримання і реакцій автентичності консервантів на прикладі уротропіну.
Реактиви: формалін, NH4OH, NaOH, AgNO3, саліцилова кислота.
Уротропін (Е 239) застосовується для консервування обмеженої кількості продуктів, у Росії – для консервування ікри лососевих риб. ДСД – 0,15 мг/кг.
Метод отримання
Методика роботи. У конічну колбу вносять 50 мл 40%-го розчину формаліну та приливають при охолодженні (вода з кригою) малими порціями 100 мл 10 %-го розчину аміаку. Ця реакція є екзотермічною, тому температура не повинна перевищувати 15–18°С. Потім колбу закривають пробкою та залишають на 6–8 год. Протягом цього часу аміак повинен бути в надлишку (відчувається запах аміаку, реакція лужна за фенолфталеїном). Після цього рідину упарюють (без кипіння) на водяній бані до появи значної кількості кристалів. Продукт фільтрують та сушать при температурі 30–35°С.
Вихід продукту складає 70%.
Реакції на автентичність
Для якісного визначення уротропіну використовують реакцію його гідролізу в кислому середовищі та визначають наявність продуктів гідролізу.
(CH2)6N4 + 6H2O + 2H2SO4 ® 6CH2O↑ + 2(NH4)2SO4.
Методика роботи. 10 мл розчину уротропіну (1:10) та 10 мл розбавленої сульфатної кислоти вливають у термостійку колбу. Колбу занурюють у киплячу водяну баню на 2–3 хв. З’являється характерний запах формальдегіду. Отриманий розчин (розчин 1) слід поділити на декілька частин для ідентифікації продуктів гідролізу.
1. Визначення сульфату амонію
(NH4)2SO4 + 2NaOH ® 2NH3↑ + Na2SO4 + 2H2O.
Методика роботи. До частини розчину 1 додають 10 мл 30%-го розчину їдкого натру та нагрівають ще раз протягом 1–2 хв. Аміак, який виділяється, визначають за запахом або за допомогою вологого лакмусового паперу (синій колір).
2. Визначення формальдегіду
Утворений формальдегід можна виявляти різними реактивами:
2.1. Нітрат срібла
Методика роботи. До 2 мл розчину нітрату срібла додають 10–12 крапель розчину аміаку та 2–3 краплі розчину 1. Нагрівають на водяній бані при температурі 50–60ºС. Утворюється металічне срібло у вигляді дзеркала або сірого осаду.

2.2. Саліцилова кислота з утворенням ауринового барвника
 |
Методика роботи. До розчину 0,02–0,03 г саліцилової кислоти в 5 мл концентрованої сірчаної кислоти додають 2 краплі розчину 1. При нагріванні з’являється фіолетово-червоне забарвлення.
Контрольні запитання
1. З якою метою застосовуються консерванти в харчових продуктах? Які вимоги до них висуваються?
2. Які існують промислові методи отримання уротропіну?
3. Наведіть якісні реакції визначення уротропіну.
Лабораторна робота 6
Визначення кількісного вмісту консервантів
Мета роботи: провести кількісний аналіз субстанції уротропіну.
Реактиви: уротропін, H2SO4, NaOH, індикатор метиловий червоний.
Для визначення кількісного вмісту уротропіну проводять його розкладання надлишком титрованого розчину сірчаної кислоти, з подальшим титруванням залишку кислоти лугом з використанням метиленового червоного:
(CH2)6N4 + 6H2O + H2SO4 ® CH2O↑ + 2(NH4)2SO4;
H2SO4 + 2NaOH ® Na2SO4 + 2H2O.
Методика роботи. Наважку уротропіну 0,12 г розчиняють у конічній колбі в 10 мл води, приливають 50 мл 0,1 н. розчину сульфатної кислоти, суміш повільно кип’ятять протягом 30 хв та охолоджують. До охолодженого розчину приливають дві краплі розчину метилового червоного і надлишок сульфатної кислоти титрують 0,1 н. розчином їдкого натрію до переходу рожевого забарвлення в жовте.
Вміст уротропіну розраховують за формулою:

де а – наважка продукту, г.
Контрольні запитання
1. Чому харчова добавка Е 239 заборонена для використання у більшості країн світу? Для яких продуктів вона дозволена для використання в Україні та Росії?
2. Які існують методи кількісного визначення вмісту уротропіну в продуктах харчування?
3. Наведіть хімічні реакції, що використовуються в методі кислотно-основного визначення уротропіну. Який індикатор при цьому використовується і чому?
Лабораторна робота 7
Методи отримання та визначення вмісту біостимуляторів
Мета роботи: отримати кофеїн з сухого листя чаю, провести кількісне визначення.
Реактиви: кофеїн, H2SO4, КI3, Na2S2O3, крохмаль.

| C8H10O2N4 | М.м. 194,20 |
Кофеїн (1, 3, 7-триметил-2,6-діоксипурин) – одна з найважливіших похідних пурину. Його вміст в листі чаю досягає 3%, у зернах кофе – 1,5%.
Властивості. Кофеїн – білі голчасті кристали, гіркі на смак.
Розчинність. Повільно розчиняється у воді, легко розчиняється в гарячій воді та хлороформі, погано розчиняється в спирті.
Метод отримання
Возгонка кофеїну з сухого чаю. Невелику кількість розтертого чаю вміщують у порцелянову чашку і накривають її часовим склом. Чашку обережно нагрівають на плитці та спостерігають возгонку кофеїну. Він осаджується на склі у вигляді довгих, злегка забарвлених голок.
Кількісне визначення
Метод заснований на тому, що йод з йодидом калію осаджує з кислих розчинів осад складу C8H10N4×HJ×J4; надлишок йоду відтитровують розчином тіосульфату натрію.

J2 + 2Na2S2O3 ® Na2S4O6 + 2NaJ.
Методика роботи. Точну наважку 0,05–0,06 г кофеїну розчиняють в 10 мл води в мірній колбі ємністю 50 мл. До розчину додають 5 мл розведеної сульфатної кислоти, 20 мл 0,1 н. розчину йоду при перемішуванні та доводять рідину до риски. Суміш збовтують, дають осаду відстоятися, після чого відфільтровують, перші порції фільтрату відкидають. У колбу для титрування відбирають 20 мл фільтрату і титрують надлишок йоду 0,1 н. розчином тіосульфату натрію з використанням індикатора крохмалю.
Вміст кофеїну розраховують за формулою:
 ,
,
де Vк – об’єм мірної колби, мл;
Vп – об’єм проби фільтрату, взятої для титрування, мл;
a – маса наважки, г.
Контрольні запитання
1. Які існують методи отримання кофеїну з рослинної сировини і шляхом хімічного синтезу?
2. Які існують методи визначення кількісного вмісту кофеїну в продуктах харчування?
3. На чому заснований йодометричний метод кількісного визначення кофеїну? Наведіть реакції.
Лабораторна робота № 8
Методи отримання харчових барвників
Мета роботи: отримати хлорофіли з рослинної сировини.
Хлорофіли – це магнійзаміщені похідні порфірину – природні пігменти, які надають зеленого забарвлення багатьом рослинам, овочам і плодам.
Хлорофіл (Е 141) – зелений пігмент рослин – складається із суміші декількох пігментів: синьо-зеленого хлорофілу a і жовто-зеленого хлорофілу b, які містяться у співвідношенні 3:1. При пропусканні розчину хлорофілу через колонку відбувається сорбція і поділ пігментів відповідно до їх вибіркової сорбції.
Хроматографічне розділення пігментів зеленого листя рослин складається з наступних етапів:
1) екстрагування пігментів з рослинного матеріалу;
2) утворення хроматограми;
3) одержання розчинів хлорофілу-a, хлорофілу-b і каротиноїдів.

При підготовці розчину для хроматографування пігменти вилучають із рослинного матеріалу полярним розчинником, а потім розчиняють у суміші неполярних розчинників. Отриманий розчин хроматографують. Первинну хроматограму промивають полярним розчинником і в розчині, що витікає, збирають окремі фракції пігментів.
Методика визначення. Для аналізу беруть наважку рослинного матеріалу зі свіжого зеленого (3–5 г) або сухого листя (1 г). Свіжий рослинний матеріал поміщають у порцелянову ступку і ретельно розтирають. Для кращого розтирання додають дрібне скло або кварцовий пісок. Якщо для досліду взято сухий матеріал, то його подрібнюють у ступці.
В отриману рослинну масу додають 15 мл суміші бензину і бензолу (9:1) і 10 мл ацетону, знову розтирають та перемішують. Суміш переносять на скляний фільтр № 2. Ступку очищають, обполіскуючи її чистим ацетоном. Промивну рідину також переносять на фільтр № 2. Екстракт пігментів відфільтровують. Для більш повного відділення пігментів масу, що залишилася на фільтрі, промивають декількома порціями ацетону (по 5 мл) доти, доки фільтрат, що витікає, не стане безбарвним, тобто не буде містити пігментів.
Отриманий після фільтрування екстракт пігментів переносять із приймача в ділильну лійку, відокремлюють ацетон шляхом обережного введення в лійку 50 мл дистильованої води. Рідину в ділильній лійці злегка збовтують. Слід уникати сильного збовтування, тому що може утворитися стійка емульсія.
Після того, як відбудеться розшарування двох рідких фаз, що не змішуються, нижню фазу (вода – ацетон) зливають. У ділильну лійку обережно вводять нову порцію води (50 мл). Після розшарування рідин водну фазу зливають. Операцію відмивання ацетону проводять 10 разів. Бензино-бензольний екстракт просушують після видалення всіх водорозчинних речовин. Для цього екстракт переносять у колбу ємністю 50 мл, у яку додають 2–3 г прожареного сульфату натрію. Екстракт відфільтровують через скляний або паперовий фільтр.
Хроматографування проводять у скляній колонці розміром 20´1 см. Унизу колонки поміщають ватний тампон і вносять невеликими порціями сорбент CaCO3 або цукровий порошок. Ущільнення сорбенту проводять постукуванням трубки об тверду поверхню.
У підготовлену колонку вводять порцію (5 мл) екстракту. Отриману первинну хроматограму промивають сумішшю бензину й бензолу (9:1). При промиванні колонки відбувається поділ зон, угорі колонки утвориться зелена зона b-хлорофілу, потім синьо-зелена a-хлорофілу, трохи нижче розташовується зона жовтого кольору (каротиноїди).
При подальшому промиванні колонки розчинником у фільтрат переводяться спочатку жовті пігменти (каротиноїди), потім a-хлорофіл й b-хлорофіл. Фракції збирають в окремі приймачі.
Контрольні запитання
1. Які групи барвників застосовуються в харчовому виробництві? У чому переваги і недоліки природних та синтетичних барвників?
2. Наведіть структурну формулу хлорофілу.
3. Яким чином розділяють природні барвники при їх виділенні з рослинної сировини?
Лабораторна робота № 9
Методи отримання й ідентифікації харчових танінів
Мета роботи: отримати таніни з рослинної сировини, провести якісне визначення.
Реактиви: желатин, NaCl, кофеїн, K2Cr2O7, H3PMo12O40, H3PW12O40, залізоамонійні галуни, бромна вода, Pb(CH3COO)2, Рb(CH3COO)2·Рb(ОН)2, NaNO2, формальдегід, HCl, ванілін.