Митохондрии и митохондриальные болезни: краткие сведения
Шершевского-Тернера
Четкой связи возникновения синдрома Тернера с возрастом и какими-либо заболеваниями родителей не выявлено. Однако беременности обычно осложняются токсикозом, угрозой выкидыша, а роды часто бывают преждевременными и патологическими. Особенности беременностей и родов, заканчивающихся рождением ребенка с синдромом Тернера, — следствие хромосомной патологии плода. Нарушение формирования половых желез при синдроме Тернера обусловлено отсутствием или структурными дефектами одной половой хромосомы (X-хромосомы).
У эмбриона первичные половые клетки закладываются почти в нормальном количестве, но во второй половине беременности происходит их быстрая инволюция (обратное развитие), и к моменту рождения ребенка количество фолликулов в яичнике по сравнению с нормой резко уменьшено или они полностью отсутствуют. Это приводит к выраженной недостаточности женских половых гормонов, половому недоразвитию, у большинства больных — к первичной аменорее (отсутствию менструаций) и бесплодию. Возникшие хромосомные нарушения являются причиной возникновения пороков развития. Возможно также, что сопутствующие аутосомные мутации играют определенную роль в появлении пороков развития, поскольку существуют состояния, сходные с синдромом Тернера, но без видимой хромосомной патологии и полового недоразвития.
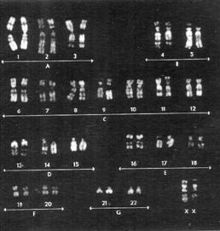

Кариотип 45,(X0)=70% / 46,(XX)=30% — мозаичная форма синдрома Тернера.
При синдроме Тернера половые железы обычно представляют собой недифференцированные соединительнотканные тяжи, не содержащие элементов гонад. Реже встречаются рудименты яичников и элементы яичек, а также рудименты семявыносящего протока. Другие патологические данные соответствуют особенностям клинических проявлений. Наиболее важны изменения костно-суставной системы — укорочение пястных и плюсневых костей, аплазия (отсутствие) фаланг пальцев, деформация лучезапястного сустава, остеопороз позвонков. Рентгенологически при синдроме Тернера турецкое седло и кости свода черепа обычно не изменены. Отмечаются пороки сердца и крупных сосудов (коарктация аорты, незаращение боталлова протока, незаращение межжелудочковой перегородки, сужение устья аорты), пороки развития почек. Проявляются рецессивные гены дальтонизма и других заболеваний.
Синдром Шерешевского-Тёрнера встречается много реже, чем трисомия Х, синдром Клайнфельтера (ХХУ, ХХХУ), а также ХУУ, что указывает на наличие сильного отбора против гамет, не содержащих половых хромосом, или против зигот ХО. Это предположение подтверждается достаточно часто наблюдемой моносомией Х среди спонтанно абортированных зародышей. В связи с этим допускается, что выжившие зиготы ХО являются результатом не мейотического, а митотического нерасхождения, или утраты X-хромосомы на ранних стадиях развития. Моносомии УО у человека не обнаружено. Популяционная частота 1:1500
Синдром Клайнфельтера встречается у 1 из 500 мальчиков. Больные с классическим вариантом синдрома имеют кариотип 47,XXY . Возможны и другие кариотипы, а у 10% больных выявляется мозаицизм 46,XY/47,XXY, встречаются и более редкие кариотипы: 48,XXXY ; 49,XXXXY ;48,XXYY ; 49,XXXYY . Синдром обычно проявляется в подростковом возрасте как задержка полового развития. Половой член и яички уменьшены , телосложение евнухоидное , имеютсягинекомастия и умеренная задержка психического развития . Больные предрасположены ксахарному диабету , заболеваниям щитовидной железы и раку молочной железы . Наличие в кариотипе не менее двух Х-хромосом и одной Y-хромосомы - самая распространенная причина первичного гипогонадизма у мужчин.
Примерно у 10% больных с синдромом Клайнфельтера наблюдается мозаицизм 46,XY/47,XXY. Поскольку в формировании фенотипа участвует клон клеток с нормальным кариотипом, больные с мозаицизмом 46,XY/47,XXY могут иметь нормально развитые половые железы и быть фертильными. Добавочная Х-хромосома в 60% случаев наследуется от матери, особенно припоздней беременности . Риск наследования отцовской Х-хромосомы не зависит от возраста отца.
Для синдрома Клайнфельтера характерен фенотипический полиморфизм. Наиболее частые признаки: высокорослость , непропорционально длинные ноги , евнухоидное телосложение , маленькие яички (длинная ось менее 2 см). Производные вольфова протока формируются нормально. В детском возрасте нарушения развития яичек незаметны и могут не выявляться даже при биопсии. Эти нарушения обнаруживают в пубертатном периоде и позднее. В типичных случаях при биопсии яичка у взрослых находят гиалиноз извитых семенных канальцев,гиперплазию клеток Лейдига , уменьшение численности или отсутствие клеток Сертоли ; сперматогенез отсутствует. Больные, как правило, бесплодны (даже если есть признаки сперматогенеза). Формирование вторичных половых признаков обычно нарушено: оволосение лица и подмышечных впадин скудное или отсутствует; наблюдается гинекомастия; отложение жира и рост волос на лобке по женскому типу. Как правило, психическое развитие задерживается, но у взрослых нарушения интеллекта незначительны. Нередко встречаются нарушения поведения, эпилептическая активность на ЭЭГ, эпилептические припадки. Сопутствующие заболевания: рак молочной железы , сахарный диабет , болезни щитовидной железы , хронические обструктивные заболевания легких .
Способы лечения бесплодия при синдроме Клайнфельтера пока не разработаны. Заместительную терапию тестостероном обычно начинают с 11-14 лет; при дефиците андрогенов она существенно ускоряет формирование вторичных половых признаков. У взрослых больных на фоне лечения тестостероном повышается половое влечение. При гинекомастии может потребоваться хирургическое вмешательство. Психотерапия способствует социальной адаптации больных с синдромом Клайнфельтера и больных с другими аномалиями половых хромосом.
Синдром Клайнфельтера обычно проявляется гипоплазией яичек , бесплодием , гинекомастией ,гипогонадизмом ; иногда наблюдаются умеренная умственная отсталость и асоциальное поведение . Для синдрома Клайнфельтера характерен кариотип 47,XXY . Такая трисомия нарушает жизнеспособность половых клеток и обусловливает атрофию семенных канальцев иазооспермию . Иногда гипоплазия яичек является единственным признаком заболевания у внешне здоровых мужчин. Синдром Клайнфельтера (полисемия по Х-хромосоме у мужчин -кариотип 47,XXY и другие варианты), предрасполагают к развитию острых миелоидных лейкозов
синдром Патау ( трисомия по 13-й хромосоме ), предрасполагают к развитию острых миелоидных лейкозов .
Синдром Патау (СП) выделен в самостоятельную нозологическую форму в 1960 г. в результате генетического исследования, проведенного у детей с врожденными пороками развития.
Трисомия по 13-й хромосоме ( синдром Патау ) обнаруживается у новорожденных с частотой около 1:5000 - 1:7000 и связана с широким спектром пороков развития. Наиболее распространенные дефекты: микрофтальмия или анофтальмия , расщелины верхней губы и твердого неба , наличие непарной резцовой кости , голопрозэнцефалия . Эндокринные нарушения: гипопитуитаризм , гетеротопия поджелудочной железы , гипоплазия наружных половых органов .
Цитогенетические варианты этого синдрома следующие. Простая полная трисомия-13 как следствие нерасхождение хромосом в мейозе у одного из родителей (главным образом у матери) встречаются у 80 - 85% больных. Остальные случаи обусловлены в основном передачей дополнительной хромосомы (точнее ее длинного плеча) в робертсоновских транслокациях типа D/13 и G/13. Другие цитогенетические варианты ( мозаицизм , изохромосома , неробертсоновские транслокации) обнаружены, но они встречаются крайне редко. Клиническая и патологоанатомическая картины простых трисомных форм и транслакоционных не различается.
Соотношение полов при СП близко к 1:1. Дети с СП рождаются с истинной пренатальной гипоплазией (на 25 - 30% ниже средних величин), которую нельзя объяснить небольшой недоношенностью (средний срок беременности 38,3 нед). Характерным осложнением беременности при вынашивании плода с СП является многоводие: оно встречается почти в 50% случаев СП.
Для СП характерны множественные врожденные пороки развития головного мозга и лица. Это патогенетическая единая группа ранних (и, следовательно, тяжелых) нарушений формирования головного мозга, глазных яблок, костей мозговой и лицевой частей черепа. Окружность черепа обычно уменьшена, встречается и тригоноцефалия. Лоб скошенный, низкий; глазные щели узкие, переносье запавшее, ушные раковины низко расположенные и деформированные. Типичный признак СП - это расщелины верхней губы и неба (обычно двухсторонние). Всегда обнаруживаются пороки нескольких внутренних органов в разной комбинации: дефекты перегородок сердца, незавершенный поворот кишечника, кисты почек, аномалии внутренних половых органов, дефекты поджелудочной железы. Как правило, наблюдается полидактилия (чаще двухсторонняя и на руках) и флексорное положение кистей. Частота встречаемости разных симптомов у детей с СП представлена в табл. 5.3 Клиническая диагностика СП основывается на сочетании характерных пороков развития. При подозрении на СП показано ультразвуковое исследование всех внутренних органов.
В связи с тяжелыми врожденными пороками развития большинство детей с СП умирают в первые недели или месяцы (95% - до 1 года). Однако некоторые больные живут в течение нескольких лет. Более того, в развитых странах отмечаются тенденция увеличения продолжительности жизни больных СП до 5 лет (около 15% детей) и даже до 10 лет (2 - 3% детей). Другие синдромы врожденных пороков развития ( синдромы Меккеля и Мора , тригоноцефалия Опитца ) по отдельным признакам совпадают с СП. Решающим фактором в диагностике является исследование хромосом. Цитогенетическое исследование показано во всех случаях, в том числе у умерших детей. Точный цитогенетический диагноз необходим для прогноза здоровья будущих детей. ( трисомия по 13-й хромосоме ), предрасполагают к развитию острых миелоидных лейкозов .
Синдром Патау (СП) выделен в самостоятельную нозологическую форму в 1960 г. в результате генетического исследования, проведенного у детей с врожденными пороками развития.
Трисомия по 13-й хромосоме ( синдром Патау ) обнаруживается у новорожденных с частотой около 1:5000 - 1:7000 и связана с широким спектром пороков развития. Наиболее распространенные дефекты: микрофтальмия или анофтальмия , расщелины верхней губы итвердого неба , наличие непарной резцовой кости , голопрозэнцефалия . Эндокринные нарушения: гипопитуитаризм , гетеротопия поджелудочной железы , гипоплазия наружных половых органов .
Цитогенетические варианты этого синдрома следующие. Простая полная трисомия-13 как следствие нерасхождение хромосом в мейозе у одного из родителей (главным образом у матери) встречаются у 80 - 85% больных. Остальные случаи обусловлены в основном передачей дополнительной хромосомы (точнее ее длинного плеча) в робертсоновских транслокациях типа D/13 и G/13. Другие цитогенетические варианты ( мозаицизм , изохромосома , неробертсоновские транслокации) обнаружены, но они встречаются крайне редко. Клиническая и патологоанатомическая картины простых трисомных форм и транслакоционных не различается.
Соотношение полов при СП близко к 1:1. Дети с СП рождаются с истинной пренатальной гипоплазией (на 25 - 30% ниже средних величин), которую нельзя объяснить небольшой недоношенностью (средний срок беременности 38,3 нед). Характерным осложнением беременности при вынашивании плода с СП является многоводие: оно встречается почти в 50% случаев СП.
Для СП характерны множественные врожденные пороки развития головного мозга и лица. Это патогенетическая единая группа ранних (и, следовательно, тяжелых) нарушений формирования головного мозга, глазных яблок, костей мозговой и лицевой частей черепа. Окружность черепа обычно уменьшена, встречается и тригоноцефалия. Лоб скошенный, низкий; глазные щели узкие, переносье запавшее, ушные раковины низко расположенные и деформированные. Типичный признак СП - это расщелины верхней губы и неба (обычно двухсторонние). Всегда обнаруживаются пороки нескольких внутренних органов в разной комбинации: дефекты перегородок сердца, незавершенный поворот кишечника, кисты почек, аномалии внутренних половых органов, дефекты поджелудочной железы. Как правило, наблюдается полидактилия (чаще двухсторонняя и на руках) и флексорное положение кистей. Частота встречаемости разных симптомов у детей с СП представлена в табл. 5.3 Клиническая диагностика СП основывается на сочетании характерных пороков развития. При подозрении на СП показано ультразвуковое исследование всех внутренних органов.
В связи с тяжелыми врожденными пороками развития большинство детей с СП умирают в первые недели или месяцы (95% - до 1 года). Однако некоторые больные живут в течение нескольких лет. Более того, в развитых странах отмечаются тенденция увеличения продолжительности жизни больных СП до 5 лет (около 15% детей) и даже до 10 лет (2 - 3% детей). Другие синдромы врожденных пороков развития ( синдромы Меккеля и Мора , тригоноцефалия Опитца ) по отдельным признакам совпадают с СП. Решающим фактором в диагностике является исследование хромосом. Цитогенетическое исследование показано во всех случаях, в том числе у умерших детей. Точный цитогенетический диагноз необходим для прогноза здоровья будущих детей.
Синдром Эдвардса (трисомия по 18 хромосоме) )
Трисомия по 18-й хромосоме (синдром Эдвардса) встречается у новорожденных с частотой от 1:3300 до 1:10000; у девочек бывает в 3 раза чаще, чем у мальчиков. Больные дети часто рождаются недоношенными или переношенными. Нарушения при трисомии по 18-й хромосоме гораздо тяжелее, чем при синдроме Дауна ; лишь 50% пробандов доживают до 2-месячного возраста; 10% живут 1 год. Средняя продолжительность жизни мальчиков - 60, девочек - 280 дней. Клиническая картина: череп необычной формы ( узкий лоб и широкий выступающий затылок ), низкое расположение ушей , микрогнатия , сгибательная контрактура кистей и стоп , дисплазия стоп , пороки сердца , сильная задержка психического развития . Главные нарушения обмена веществ и эндокринные расстройства: гипоплазия подкожной клетчатки, сильная задержка роста . Дисгенезия щитовидной железы или надпочечников встречается менее чем у 10% больных.
Синдром Дауна (СД), трисомия-21 , - наиболее изученная хромосомная болезнь. Описана врачом в 1866 г. и названа им монголизмом. [ The Metabolic Basis of Inherited Disease,1983 ]. Частота СД среди новорожденных равна 1:700 - 1:800 , не имеет какой-либо временной, этнической или географической разницы при сравнении одинакового возраста родителей. Частота рождения детей с СД зависит от возраста матери и в меньшей мере от возраста отца.[ Мюнтцинг А.,1967 ]. Генетические дефекты, лежащие в основе синдрома Дауна, - самая частая причина врожденных пороков развития и умственной отсталости. Распространенность синдрома Дауна велика: он обнаруживается у 1 из 670 новорожденных. Примерно в 94% случаев синдром обусловлен трисомией по 21-й хромосоме . У 3% больных наблюдается мозаицизм. В остальных случаях синдром вызван спорадической или наследуемой транслокацией 21-й хромосомы . Как правило, такие транслокации возникают в результате слияния центромеры 21-й хромосомы и другой акроцентрической хромосомы. Фенотип больных определяется трисомией 21q22. Повторный риск рождения ребенка с синдромом Дауна у родителей с нормальным кариотипом составляет около 1%. Повторный риск у лиц с мозаицизмом и носителей сбалансированной транслокации существенно выше. Пожилой возраст матери - единственный фактор риска, для которого четко установлена связь с синдромом Дауна
Цитогенетические варианты СД разнообразны. Однако основную долю (94 - 95%) составляют случаи простой полной трисомии-21 как следствие нерасхождения хромосом в мейозе . При этом вклад материнского нерасхождения в эти гаметические формы болезни составляет 80%, а отцовского - только 20%. Причины такой разницы неясны. Небольшая (около 2%) доля детей с СД имеют мозаичные формы(47+21/46). Примерно 4% больных СД имеют транслокационную форму трисомии по типу робертсоновских транслакаций между акроцентриками (D/21 и G/21). Почти 50% транслокационных форм наследуется от родителей-носителей, и 50% - это транслокации.
Синдром "кошачьего крика"
Синдром Лежена - другие названия: синдром 5р-, синдром "кошачьего крика", - проявляющийся умственной отсталостью, характерными чертами лица, в грудном возрасте плачем, напоминающим мяуканье.
Хромосома половая: трисомия по Х-хромосоме (кариотип 47,ХХХ)
Трисомия по Х-хромосоме ( 47,ХХХ ) встречается у новорожденных девочек с частотой 1:1000; редко диагностируется в раннем детстве; взрослые больные обычно имеют нормальный женский фенотип.
Немногочисленные проспективные исследования показали, что у женщин с кариотипом 47,ХХХ наиболее часто отмечаются: высокий рост ; умственная отсталость (как правило, легкой степени); позднее развитие речи ; эпилепсия ; дисменорея ; бесплодие . Риск рождения ребенка с трисомией по Х-хромосоме повышен у пожилых матерей . Для фертильных женщин с кариотипом 47,ХХХ риск рождения ребенка с таким же кариотипом невелик. По-видимому, существует защитный механизм, предотвращающий образование или выживание анеуплоидных гамет или зигот.
При полисомии Х-хромосомы с числом Х-хромосом более трех (например, кариотип 48,ХХХХ , кариотип 49,ХХХХХ) высока вероятность тяжелой умственной отсталости , нарушения пропорций лица , пороков развития скелета или пороков внутренних органов . Синдромы такого рода встречаются редко и обычно имеют спорадический характер.
41,Гены-триггеры мультифакториальных заболеваний: введение
Поломки регуляторных генетических механизмов, приводящие к тяжелым мультифакториальным заболеваниям, могут быть спровоцированы не только функционально неполноценными аллелями " генов внешней среды ", но и мутациями в структурных генах или генах, обеспечивающих клеточный гомеостаз . Для удобства изложения эту достаточно полиморфную группу генов мы называем генами- триггерами мультифакториальных болезней. К ним можно отнести онкогены , многие из которых играют решающую роль в возникновении опухолей, а также гены супрессоры (например ген р53 ), мутации в которых приводят к активации соответствующих онкогенов и включению цепи метаболических реакций, вызывающих в конечном счете трансформацию клеток. Для многих десятков таких генов уже известны неблагоприятные аллельные варианты предрасполагающие к заболеваниям. Число уже известных генов-триггеров мультифакториальных заболеваний сегодня достигло 50 и быстро увеличивается. Только в последние годы идентифицированы мутантные аллели гена СС16 , предрасполагающие в гомозиготном состоянии к астме (10% населения); мутации в гене Фактора V свертывания крови, резко увеличивающие вероятность тромбозов ; наконец, аллельные полиморфизмы гена TGF2 , коррелирующие с такими достаточно частыми аномалиями внутриутробного развития, как расщелина верхней губы ( заячья губа ) и незаращение твердого неба ( волчья пасть ). Однако сравнительно частые мутации структурных генов далеко не всегда вредны для организма. Иногда, достаточно редко, они могут оказаться даже полезными. Обычно для иллюстрации данного положения ссылались на пример серповидноклеточной анемии в странах Средиземноморья, где мутации в глобиновых генах в гетерозиготном состоянии служат действенной защитой от малярии, тогда как в гомозиготном - оказываются летальными. Подобный сбалансированный полиморфизм недавно описан и для гена муковисцидоза (CFTR) . Экспериментально доказано, что особи, гетерозиготные по мутациям гена CFTR, обладают повышенной устойчивостью к холерному токсину . Вероятно, именно этим можно объяснить столь высокую частоту мутаций гена CFTR в популяции, несмотря на то, что в гомозиготном состоянии они приводят к тяжелому наследственному заболеванию.
Импринтинг геномный
Изменения одиночных генов или целых районов хромосом родителей при мейозе могут приводить к появлению гамет с генетическими дефектами. В таких случаях фенотипическое проявление дефекта у ребенка зависит от того, какая именно гамета участвует в образовании зиготы.
Характер проявления аутосомно-доминантных болезней зависит от происхождения дефектного аллеля. Например, ювенильная форма болезни Гентингтона наблюдается только у детей больных отцов, а наследственная атрофическая миотония - только у детей больных матерей.
Импринтинг наблюдается у многих больных с хромосомными делециями. При синдроме Прадера-Вилли всегда обнаруживается делеция отцовской 15-й хромосомы 15q11-13. Делеция этого же района материнской 15-й хромосомы обусловливает развитие синдрома Эйнджелмена (комплекс врожденных психических расстройств) . Синдром Эйнджелмена легко отличим от синдрома Прадера-Вилли.
Роль однородительской дисомии в патологии во многом усугубляется геномным импринтингом, который приводит к неодинаковой экспрессии материнской и отцовской копий гена.
У мыши и человека отцовские копии гена ИФР-II (IGF2) и гена полипептида малого ядерного рибонуклеопротеида N ( SNRPN ) экспрессируются, а материнские - нет, и наоборот, материнская копия гена H19 экспрессируется, а отцовская - нет. Ген (или гены), вызывающий синдром Прадера-Вилли , по-видимому, экспрессируется с отцовской копии, а ген, вызывающий синдром Ангельмана , - с материнской.
Характер наследования болезней при геномном импринтинге отличается от обычного менделевского.
Если в результате геномного импринтинга экспрессируется только один аллель аутосомного гена, а экспрессия другого аллеля подавлена, то этот ген функционально гемизиготен . Хотя в таком случае признак нельзя рассматривать как доминантный, но болезнь, как и в случае доминантного наследования, проявляется в нескольких поколениях ( вертикальное наследование ). Описаны большие родословные, в которых геномный импринтинг влияет на наследование болезни.
Как показано на рис. 65.18 , семейная параганглиома проявляется только при наследовании мутантного аллеля от отца. Напротив, синдром Ангельмана проявляется только при наследовании мутантного аллеля от матери.
МИТОХОНДРИАЛЬНЫЕ БОЛЕЗНИ
Комплексы дыхательной цепи и окислительное фосфорилирование
Синдром Кирнса-Сейра
Синдром MERRF
Синдром MELAS
Болезнь Ли (подострая некротическая энцефаломиопатия)
Синдром Лебера
Митохондрии и митохондриальные болезни: краткие сведения
В 1972 г. Ольсон с соавт. с помощью модифицированной трехцветной окраски обнаружили в поперечнополосатых мышцах волокна с аномально большим количеством митохондрий. Они предложили новый термин - " рваные мышечные волокна ". При электронной микроскопии видно, что митохондрии в таких волокнах увеличены , часто имеют необычную форму и содержат кристаллические включения . Со времени этих исследований представления о митохондриальных миопатиях и наследственных митохондриальных болезнях вообще существенно расширились.
Митохондриям принадлежит ведущая роль в образовании энергии. В результате окисления углеводов, жиров и белков образуются восстановительные эквиваленты (электроны и атомы водорода), которые переносятся по дыхательной цепи. Высвобождающаяся при этом энергия переходит в энергию электрохимического градиента для протонов на внутренней мембране митохондрий, а та, в свою очередь, используется для синтеза АТФ . Этот процесс называется окислительным фосфорилированием .
Генетические особенности митохондрий необычны. Каждая митохондрия обладает набором генов, отличающимся от ядерных. Митохондриальная ДНК человека представляет собой двухцепочечную кольцевую молекулу, содержащую 16569 пар оснований. Она кодирует 22 молекулы тРНК, 2 - рРНК и 13 полипептидов ферментов дыхательной цепи. Столь же своеобразно наследуются и митохондриальные болезни. Митохондриальная ДНК передается с цитоплазмой гамет, причем почти исключительно яйцеклеток , поскольку сперматозоиды практически не содержат митохондрий. Поэтому митохондриальные гены - и митохондриальные болезни - наследуются исключительно по материнской линии.
Многие мутации нарушают функции митохондрий.
Митохондриальный геном состоит из кольцевой ДНК размером 16600 нуклеотидов, которая кодирует рРНК и тРНК, участвующие в митохондриальной системе трансляции, и некоторые белки, необходимые для окислительного фосфорилирования. Многие митохондриальные белки кодируются генами ядерного генома, синтезируются в цитоплазме и затем транспортируются в митохондрии. Поэтому мутации, нарушающие функции митохондрий, могут происходить как в митохондриальном, так и в ядерном геномах.
Мутации в ядерном геноме наследуются так же, как и другие ядерные гены, а наследование мутаций в митохондриальном геноме носит иной характер.
Число митохондрий в клетке непостоянно, как и количество митохондриальных геномов в митохондрии (обычно от 2 до 10), так что каждая клетка содержит тысячи копий митохондриальной ДНК. Эта ДНК передается через яйцеклетку в количестве 200000-300000 копий и в незначительной степени через сперматозоиды . Это означает, что все люди наследуют митохондриальные геномы от матери.
Если митохондриальный ген несет патологическую мутацию, она обычно представлена только в части митохондриальных геномов в клетке. Эту гетерогенность митохондриальных геномов в клетке или в организме называют гетероплазмией .
Мутацию в митохондриальном геноме женщины унаследуют все ее дети, а мужчина с такой мутацией, наоборот, не передаст ее детям. Это приводит к типичному вертикальному наследованию , когда больная женщина передает заболевание большинству своих детей.
Соотношение митохондрий с мутантными и нормальными геномами у представителей одной семьи непостоянно, что приводит к фенотипической гетерогенности.
Мутации в митохондриальном геноме обусловливают: синдром Кирнса-Сейра , синдром Лебера ( наследственную атрофию зрительных нервов ), синдром MERRF ( Myoclonic Epilepsy with Ragged Red Fibres - миоклоническую эпилепсию с рваными мышечными волокнами ) и другие болезни.