Краткие теоретические сведения
ВВЕДЕНИЕ
Физико-химические методы анализа, основаны на зависимости физических свойств вещества от его природы, причем аналитический сигнал представляет собой величину физического свойства, функционально связанную с концентрацией или массой определяемого компонента. Физико-химические методы анализа могут включать химические превращения определяемого соединения, растворение образца, концентрирование анализируемого компонента, маскирование мешающих веществ и других. В отличие от «классических» химических методов анализа, где аналитическим сигналом служит масса вещества или его объем, в физико-химических методах анализа в качестве аналитического сигнала используют интенсивность излучения, силу тока, электропроводность, разность потенциалов и др.
Важное практическое значение имеют методы, основанные на исследовании испускания и поглощения электромагнитного излучения в различных областях спектра. К ним относится спектроскопия (например, люминесцентный анализ, спектральный анализ). К важным физико-химическим методам анализа принадлежат электрохимические методы, использующие измерение электрических свойств вещества (потенциометрия).
При выполнении физико-химических методов анализа используют специальную, иногда довольно сложную, измерительную аппаратуру, в связи с чем эти методы часто называют инструментальными. Многие современные приборы оснащены встроенными ЭВМ, которые позволяют находить оптимальные условия анализа (напр., спектральную область получения наиболее точных результатов при анализе смеси окрашенных веществ), выполняют расчеты и т.д.
Физико-химические методы анализа часто используют при определении низких содержаний (порядка 10-3% и менее), где классические химические методы анализа обычно неприменимы. В области средних и высоких концентраций химические и физико-химические методы анализа успешно конкурируют между собой, взаимно дополняя друг друга. Физико-химические методы анализа развиваются в направлении поиска новых химических аналитических свойств вещества, увеличения точности анализа, конструирования новых прецизионных аналитических приборов, совершенствования существующих методик и автоматизации анализа.
МЕТОДИЧЕСКИЕ УКАЗАНИЯ К ЛАБОРАТОРНЫМ ЗАНЯТИЯМ
Лабораторная работа 1
ПОТЕНЦИОМЕТРИЧЕСКОЕ ОПРЕДЕЛЕНИЕ ЩЁЛОЧНОСТИ ВОДЫ
Цель работы:
1. Ознакомиться с потенциометрическим методом.
2. Определить щёлочность водопроводной воды.
Краткие теоретические сведения
Потенциометрия основана на измерении потенциала, возникающего между электродом и раствором, величина которого зависит от количества (концентрации) или качества (природы) определяемого компонента. Такой электрод называют индикаторным. Таким образом, потенциал электрода в потенциометрии – аналитический сигнал.
Выделяют две группы методов потенциометрии, различающиеся природой используемых индикаторных электродов и электрохимической реакцией на них:
-Редоксметрия (классическая потенциометрия) с использованием электродов с электронным типом проводимости. Возникающий при этом потенциал подчиняется уравнению Нернста:
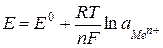 , (1.1)
, (1.1)
где 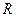 - универсальная газовая постоянная, 8,31 Дж/моль;
- универсальная газовая постоянная, 8,31 Дж/моль;  - температура, К;
- температура, К;  - число Фарадея, 96500 Кл/моль-экв;
- число Фарадея, 96500 Кл/моль-экв;  - число электронов, участвующих в редокс-процессе; aMen+ - активность потенциалопределяющих ионов.
- число электронов, участвующих в редокс-процессе; aMen+ - активность потенциалопределяющих ионов.
Электрохимическая реакция, протекающая на электроде:
Me+n + ne ® Me. (1.2)
Частный случай редоксметрии – потенциометрическое титрование.
- Ионометрия – разновидность потенциометрии, где в качестве индикаторных электродов используются электроды с ионным типом проводимости – мембранные ионселективные электроды (ИСЭ), потенциал которых возникает вследствие обмена ионами мембраны с раствором и подчиняется уравнению Никольского:
 , (1.3)
, (1.3)
где  - условный стандартный потенциал;
- условный стандартный потенциал;  - активность определяемых ионов
- активность определяемых ионов  ;
;  - активность мешающих
- активность мешающих  - ионов;
- ионов;  - коэффициент селективности.
- коэффициент селективности.
Знак «+» ставится при расчетах потенциалов катионселективных электродов, знак «-» – анионселективных.
Метод характеризуется сравнительно невысокой точностью определения. Потенциал измеряется на приборах с точностью ±1 мВ, что соответствует погрешности определения концентрации однозарядных ионов ±4%. Концентрации многозарядных ионов определяются с еще большими погрешностями. Другим ограничением является пропорциональность величины потенциала логарифму активности, а не концентрации, переход к концентрации затруднителен. Существующие приемы ионометрических измерений направлены на преодоление указанных ограничений.
Электроды в потенциометрии
Согласно электрохимическим реакциям электроды, используемые в потенциометрии, различают по роду.
Электроды 1-го рода обратимы относительно собственных ионов, а их потенциал – функция активности этих ионов в растворе. Например, серебряный электрод: Ag½Ag+. Электрохимическую реакцию, протекающую на электроде, можно представить в виде:
Ag+ + e ® Ag0 (1.4)
Электрод активен и сам принимает участие в обмене электрона.
Потенциал такого электрода подчиняется уравнению Нернста:
 (1.5)
(1.5)
К этой группе электродов относят и индифферентные электроды, например, платиновый. Платина в раствор ионов не поставляет, а выступает лишь переносчиком электронов от окислителя к восстановителю в растворе. Если платиновый электрод погрузить в раствор, содержащий окислительно-восстановительную систему
 , (1.6)
, (1.6)
то устанавливается потенциал, отражающий состояние этой системы:
 . (1.7)
. (1.7)
Электроды первого рода используют при измерении потенциала в качестве индикаторных.
Электроды 2-го рода – это металл, находящийся в равновесии с его малорастворимой солью. Например, хлорсеребряный электрод Ag|AgCl|Cl-. Электрохимическую реакцию, протекающую на электроде, можно представить в виде:
AgCl + e ® Ag0 + Cl-. (1.8)
Потенциал такого электрода зависит от активности аниона соли, хотя сами анионы при этом не окисляются и не восстанавливаются:
 (1.9)
(1.9)
Потенциалы электродов 2-го рода хорошо воспроизводимы и устойчивы. Такие электроды используют в качестве электродов сравнения.
Потенциометрическое титрование
В основе метода лежит вариант классического титрования, но не с индикаторной, а с потенциометрической индикацией точки эквивалентности с помощью индикаторного и вспомогательного электродов.
Гальванический элемент, составленный из индикаторного ионселективного электрода и вспомогательного хлорсеребряного электрода сравнения, может быть представлен схемой:
| Ag, AgCl, KCl | Исследуемый раствор | Мембрана | Стандартный раствор | KCl, AgCl, Ag |
| Вспомогательный электрод | Вспомогательный электрод | |||
| Ионселективный электрод |
Потенциометрическая ячейка для измерения потенциала с ионселективным электродом в качестве индикаторного представлена на рисунке 1.

Рис. 1.1. Схема установки для измерения потенциала с ионоселективным электродом: 1. – вспомогательный электрод ЭВЛ-1МЗ; 2 – наконечник ионселективного электрода; 3 – жидкая мембрана; 4 – магнитная мешалка; 5 - магнит
Методика потенциометрического титрования
В стакан для титрования емкостью 50-100 мл переносят пипеткой аликвотную часть испытуемого раствора. Прибавляют необходимые реагенты и погружают в раствор электроды. Если уровень раствора мал и электроды недостаточно погружены в раствор, добавляют нужное количество воды (или другого растворителя), включают мешалку и титруют, записывая показания прибора. Целесообразно первое титрование выполнить ориентировочно, прибавляя титрант порциями по 1-2 мл.
Результаты измерений помещают в таблицу, где наряду с исходными данными вносят и данные для расчетов по дифференциальным кривым (таблица 1.1).
Таблица 1.1 - Результаты потенциометрического титрования
| V, мл | E, мВ | DVi | DEi | 
| 
| 
|
Для построения интегральной кривой используются данные столбцов 1 и 2, для построения дифференциальной кривой – столбцов 1 и 5, для построения кривой второй производной – столбцов 1 и 7.
Построив график зависимости Е от объема титранта, находят положение скачка титрования и при повторном титровании вначале добавляют титрант относительно большими порциями (2-5 мл), а вблизи скачка титрант добавляют по 0,1-0,05 мл или даже по одной капле. Точке эквивалентности соответствует область максимального скачка потенциала, максимум первой производной от потенциала по объему титранта или момент резкого падения второй производной. Графическое нахождение точки эквивалентности показано на рисунке 1.2.
 |
Рисунок 2 – Виды кривых потенциометрического титрования, с помощью которых определяют эквивалентный объем титранта: а) интегральная кривая титрования; б) график первой производной от потенциала по объему; в) график второй производной
Для обнаружения точки эквивалентности в качестве индикаторного электрода используют стеклянный электрод, обратимый относительно ионов водорода.
Определение щелочности воды
Под щелочностью воды понимают сумму содержащихся в ней анионов HCO3-, CO32-, OH-. При рН<8,3 основным компонентом щелочности являются анионы HCO3-, при рН>8,3 в зависимости от рН компонентами могут быть HCO3-, CO32-, OH-. Одновременное наличие всех трех компонентов невозможно, т.к. протекает реакция кислотно-основного взаимодействия:
 . (1.10)
. (1.10)
Граничное значение рН=8,3 для наличия гидроксильных ионов обусловлено концентрацией H2CO3 и определяется уравнением:
 , (1.11)
, (1.11)
где  .
.
Так для 10-2 М растворов  – это граничное значение рН для содержания ионов HCO3- и CO32-.
– это граничное значение рН для содержания ионов HCO3- и CO32-.
Метод определения общей щелочности воды и ее компонентов основан на потенциометрическом титровании пробы воды 0,1 М раствором HCl с индикаторным стеклянным электродом и хлоридсеребряным электродом сравнения с регистрацией всех скачков рН в соответствии с протекающими реакциями:
 (1.12)
(1.12)
 (1.13)
(1.13)
 (1.14)
(1.14)
В зависимости от компонентов щелочности возможны следующие варианты титрования:
1. На кривой титрования 1 скачок:
а) начальное значение рН>8,3 – компонентами щелочности являются ионы OH-;
б) начальное значение рН<8,3 – компонентами щелочности являются ионы HCO3-.
2. На кривой титрования 2 скачка ( V1 – объем HCl в мл для первого скачка; V2 – для второго:
а) V1=V2 – компонентами щелочности являются ионы CO32-, титруемые ступенчато. Для расчета карбонатной щелочности используют объем V1+V2=2V1=2V2
б) V1>V2 – компонентами щелочности являются OH- и CO32-
Первый скачок (V1) соответствует реакциям:
(1.15)
(1.16)
второй скачок (V2):
(1.17)
Для расчета карбонатной щелочности используют объем 2V2; щелочности, обусловленной содержанием ионов OH- - объем (V1-V2).
в) V1<V2 – компонентами щелочности являются ионы CO32- и HCO3-.
Первый скачок (V1) соответствует реакции:
(1.18)
второй скачок (V2):
(1.19)
Для расчета щелочности, обусловленной присутствием карбонатов, используют объем 2V1; гидрокарбонатной – (V2-V1). Общую щелочность воды рассчитывают с учетом (V1+V2).
Приборы, реактивы
рН-метр рН-150МЛ, стеклянный электрод типа ЭС-10603, хлорсеребряный электрод типа ЭСр-10103, бюретка вместимостью 25 мл, мерные пипетки вместимостью 10 мл, мерная колба вместимостью 100 мл, сосуд для титрования, магнитная мешалка, стандартный раствор HCl, 0,1 М.
Порядок выполнения работы
Для определения щелочности пробу воды помещают в мерную колбу вместимостью 100 мл, доводят до метки дистиллированной водой и перемешивают. Мерной пипеткой вносят 10 мл анализируемого раствора в сосуд для титрования, добавляют 40 мл дистиллированной воды, погружают электроды, включают магнитную мешалку и титруют 0,1 М раствором HCl, добавляя титрант по 0,5 мл. После добавления каждой порции кислоты показаниям прибора дают установиться и записывают результаты измерения рН.
По достижении первого скачка рН титрование продолжают до второго скачка, а затем до незначительного изменения рН. По измеренным величинам рН строят кривые титрования в координатах рН-V – интегральную и DpH/DV-V – дифференциальную.
По кривым находят точки эквивалентности и определяют объемы титранта, израсходованные на титрование по первому скачку рН (V1) и по второму скачку (V2).
Щелочность воды А, моль/л рассчитывают по формуле:
 . (1.20)
. (1.20)
По характеру кривых титрования судят о качественном и количественном составе компонентов щелочности анализируемой воды.
Потенциометрическому измерению рН воды со стеклянным электродом не мешают окраска, мутность, взвеси, присутствие свободного хлора, окислителей или восстановителей, а также повышенное содержание солей в пробе.
Контрольные вопросы
1. На чем основан потенциометрический метод анализа?
2. Какая зависимость выражается уравнением Нернста? Поясните смысл входящих в него величин.
3. Что представляют собой электроды I и II рода? Приведите примеры этих электродов.
4. Какие функции выполняют индикаторные электроды и какие – электроды сравнения? Укажите требования, которые к ним предъявляются.
5. Приведите схему установки для потенциометрических измерений.
6. В чем сущность потенциометрического определения рН раствора? Какие индикаторные электроды могут быть использованы для определения рН?
7. В каких координатах строят кривые потенциометрического титрования? Чем обусловливается выбор координат?
Лабораторная работа 2
фотометрическое определение содержания Fe3+ и Cu2+
Цель работы:
1. Ознакомиться с фотометрическим методом.
2. Изучить принципы определения концентрации ионов металлов методом градуировочного графика, сравнения, добавок и дифференциального, а также по среднему значению изомолярного коэффициента погашения.
3. Найти концентрацию Fe3+ в растворе, пользуясь методами градуировочного графика, сравнения, добавок и по среднему значению молярного коэффициента погашения; провести сопоставление результатов.
4. Найти концентрацию Cu2+ в растворе, используя подходящие варианты дифференциального метода.
Краткие теоретические сведения
Фотометрический метод анализа основан на измерении эффектов взаимодействия вещества с электромагнитным излучением оптического спектрального диапазона.
В фотометрическом анализе однородная нерассеивающая система поглощает световой поток видимой, а также ультрафиолетовой и ближней инфракрасной областей спектра. При этом она возбуждается, переходя на более высокий энергетический уровень. Анализ по поглощению световой энергии молекулами и сложными ионами анализируемых веществ составляет обширную группу методов, имеющих очень широкое применение как в научно-исследовательских лабораториях, так и на производстве. Наибольшее распространение получили методы, основанные на поглощении в видимой области спектра (в интервале длин волн 400 – 800 нм). Это объясняется возможностью получения различных интенсивно окрашенных органических и неорганических соединений, пригодных для использования в данном спектральном интервале с помощью достаточно простых и недорогих приборов.
Каждая однородная система обладает способностью поглощать излучение определенной длины волны. Нагляднее всего такая способность проявляется при работе с веществами, которые характеризуются избирательным поглощением в видимой части спектра. В результате поглощения одной или нескольких длин волн из сплошного белого света система приобретает определенную окраску (т. е. цвет любого окрашенного соединения будет дополнительным к цвету поглощенного излучения).
Количественный фотометрический анализ использует математическую зависимость между поглощательными свойствами однородной нерассеивающей системы и количеством частиц, участвующих в процессе поглощения. Следовательно, светопоглощение при определенной длине волны является материальным воплощением информации об исследуемом веществе и составляет аналитический сигнал. Обычно предпочитают выбирать такие условия фотометрирования, при которых наблюдается прямолинейная зависимость светопоглощения от концентрации раствора. Однако кроме метода, основанного на построении линейного градуировочного графика, существуют и другие варианты количественного фотометрического анализа. В частности, известны методы сравнения, добавок, дифференциальный, кинетический, широко распространен анализ многокомпонентных систем.
В фотометрической практике используют различные химические реакции, приводящие к образованию соединений, которые обладают значительным светопоглощением. При этом объектом изучения могут быть растворы как неорганических, так и органических веществ. Более всего в фотометрии распространены реакции комплексообразования, окисления-восстановления и т. д. Используемые в фотометрии реакции должны непременно сопровождаться возникновением, изменением или ослаблением светопоглащения раствора и протекать избирательно, быстро, воспроизводимо. Кроме того, желательно, чтобы окраска аналитической формы определяемого вещества была устойчивой во времени и к действию света.
Любое фотометрическое определение состоит из нескольких этапов: подготовка пробы и переведение анализируемого компонента в раствор, получение его окрашенной формы, измерение светопоглощения (т. е. регистрация аналитического сигнала при условиях обеспечивающих, максимальную интенсивность); соответствующая метрологическая оценка полученных данных. Все указанные этапы одинаково важны, поскольку от правильности выполнения каждой операции и учета любого фактора, влияющего на систему, зависит конечный результат эксперимента.
Условием применения фотометрического метода для количественного определения вещества является подчинение основному закону светопоглощения Бугера – Ламберта – Бера:
 ,
,  (2.1)
(2.1)
где А – оптическая плотность (светопоглощение) раствора; Т – его пропускание; J – интенсивность излучения, прошедшего через раствор; Jo – интенсивность светового потока, падающего на раствор; e – молярный коэффициент поглощения, l – толщина светопоглощающего слоя раствора; с – концентрация испытуемого вещества в растворе.
Важным дополнением служит закон аддитивности, сущность которого заключается в независимости поглощения индивидуального вещества от наличия других компонентов, обладающих собственным светопоглощением или индифферентных к электромагнитному излучению. Таким образом, оптическая плотность смеси компонентов, подчиняющихся основному закону и не взаимодействующих между собой, при данной длине волны поглощения равна сумме оптических плотностей отдельных компонентов при этой же длине волны.
 (2.2)
(2.2)
Измерение абсолютных значений Jo и J технически затруднительно. Поэтому для оценки степени поглощения света веществом проводят сравнение интенсивностей световых потоков, проходящих через испытуемый раствор и через раствор, поглощение которого условно принято нулю (раствор сравнения). Учитывая указанное обстоятельство, следует правильно понимать физический смысл оптической плотности.
Фотометрические приборы позволяют измерять величины оптической плотности (А) или пропускания (Т,%). Теоретически величина А изменяется от нуля до бесконечности. Однако не все величины оптической плотности могут быть измерены с достаточной точностью. Оптимальное значение оптической плотности, отвечающее минимальной погрешности измерений (мин), по теоретическим расчетам равно 0,434. погрешность не превышает 2 мин, если светопоглощение укладывается в интервал 0,1–1,2.
В фотометрии в качестве приемника и преобразователя световой энергии обычно используют фотоэлементы, сила фототока которых, согласно закону фотоэффекта, пропорциональна интенсивности падающего на них излучения. Эта сила измеряется, как правило, с помощью гальванометров – микроамперметров.
На рис. 2.1 приведена принципиальная схема двухлучевого спектрофотометра.

Рис. 2.1. Принципиальная схема двухлучевого спектрофотометра
Излучение от источника света 1 делится на два пучка системой зеркал 2. Рабочий пучок проходит через кювету с образцом 3, а пучок сравнения – через компенсатор фона 4. С помощью дискомодулятора 5 пучки попеременно направляются на входную щель монохроматора 6 и через нее – на дифракционную решетку 7, которая разлагает излучение в спектр и направляет его на выходную щель 8. Монохроматическое изображение щели попадает на приемник – висмутовый болометр 9. В отсутствии исследуемого образца интенсивности рабочего пучка и пучка сравнения одинаковы, в приемнике сигналы от этих пучков вычитаются; на выходе сигнал отсутствует. При поглощении рабочего пучка исследуемым веществом на приемник попадают лучи различной интенсивности, в результате чего в приемнике возникает переменный сигнал. После усиления и преобразования сигнала производится идентификация сигнала регистрирующим устройством 10. При медленном повороте решетки щель 8 последовательно вырезает узкие участки спектра, и вычерчивается кривая зависимости пропускания от длины волны.
До применения конкретной аналитической методики с использованием фотометрических измерений должны быть проведены некоторые предварительные измерения.
Выбор оптимальной длины волны
Для выбора светофильтров обычно измеряют оптическую плотность раствора со всеми имеющимися в данном приборе фильтрами. Тот светофильтр, при котором наблюдается максимальное значение светопоглощения, и применяют при последующих измерениях. Однако в видимой части спектра выбор фильтра может осуществляться и более быстро – по окраске раствора, с учетом того, что цвет светофильтра должен быть дополнительным к цвету исследуемого вещества.
Выбор оптимальной толщины светопоглощающего слоя
К фотометрическим приборам обычно прилагается набор кювет с различной толщиной светопоглощающего слоя (от 0,5 до 5 см). Кюветы подбирают таким образом, чтобы значения оптических плотностей серии растворов укладывались в оптимальный интервал 0,1 – 1,2, соответствующий приемлемой погрешности измерений. Выбор кювет при работе в видимой области спектра производится визуально, в соответствии с интенсивностью окраски растворов. Если растворы сильно окрашены, то пользуются кюветами с малой рабочей длиной. В случае слабо окрашенных растворов применяют кюветы большой длины, а в подобранную таким образом кювету наливают раствор из середины серии и измеряют его светопоглощение при оптимальной длине волны. Если оптическая плотность составляет примерно 0,3 – 0,5, то кювета выбрана удачно. Если же светопоглощенне выходят за границы указанного диапазона, то следует соответственно увеличить или уменьшить длину кюветы.
Выбор раствора сравнения
Раствор сравнения – раствор, светопоглощение которого принимают равным нулю. Под исследуемым подразумевают любой раствор, поглощение которого измеряют относительно раствора сравнения. Раствор, содержащий неизвестную концентрацию определяемого компонента, принято называть испытуемым (анализируемым).
Как указывалось ранее, фотометрические измерения проводят относительно раствора сравнения, светопоглощение которого условно принято равным нулю. При его выборе нужноучитывать поглощение исследуемого раствора и влияние сопутствующих компонентов. Растворы сравнения могут быть различные.
1. Растворитель (например, вода, толуол и т. п.) Обычно растворители используют лишь в том случае, если нет наложения в спектрах поглощения исследуемого соединения и реагента.
2. Раствор реагента. Он применяется при наличии наложения в спектрах поглощения растворов изучаемого комплексного соединения и реагента. Если комплекс неустойчив (и потому берется большой избыток реагента), то в растворе сравнения создают ту же его концентрацию, которая взята для приготовления исследуемого соединения. Когда комплекс устойчив (и потому достаточно малого избытка реагента, но поглощение его велико), используется один из вариантов дифференциального метода измерений.
3. Раствор контрольного опыта. Его готовят в условиях приготовления эталонных растворов, но не вводят определяемый элемент. При этом исключается влияние загрязнений, присутствующих в применяемых реактивах. Такой раствор сравнения рекомендуется для определения ультрамалых количеств элементов.
4. Раствор анализируемого объекта. Он применяется в случае, когда другие присутствующие в растворе элементы не образуют соединений с реагентом, но поглощают свет при той же длине волны.
При использовании кювет необходимо соблюдать следующие правила:
– Не держать растворы в кюветах длительное время, поскольку многие химические соединения способны адсорбироваться на поверхности стекла и загрязнять стенки кювет.
– Вымыть кюветы сразу же по окончании работы. Обычно их моютконцентрированными кислотами, тщательно промывают водой, ополаскивают дистиллятом и вытирают снаружи. От органических веществ кюветы отмывают органическими растворителями.
– Не тереть во время мытья или вытирания стенки кювет чем-либо жестким, чтобы не повредить оптическую поверхность. Оптические поверхности следует оберегать от соприкосновения с посторонними предметами и друг с другом.
– Высушивать кюветы изнутри только при работе с органическими растворителями, не смешивающимися с водой. Для ускорения сушки можно последовательно ополоснуть кюветы этанолом и ацетоном.
– Ополаскивать кюветы перед заполнением небольшими порциями очередного раствора (во избежание его разбавления). При этом вначале работают с менее концентрированными растворами.
– Вытирать перед заполнением наружные стенки кювет (даже если они сухие и кажутся чистыми) обезжиренным, мягким, не оставляющим ворса материалом. Не рекомендуется для протирки использовать фильтровальную бумагу.
– Брать кюветы только за кромки и ребра, не касаясь пальцами их рабочих поверхностей.
– Не заполнять кюветы на весу. Перед заполнением ставить их на кусочек фильтровальной бумаги.
– Заливать кюветы до метки на их боковой стенке (т. е. до такого уровня, чтобы световой поток целиком проходил через раствор).
– Не допускать попадания раствора на внешние стенки кювет. В некоторых случаях в ограниченном объеме кюветы жидкость образует мениск и при этом по углам кюветы поднимается на значительную (~5 мм) высоту, иногда переползая на внешние стенки и создавая впечатление протекания кюветы,
– Устанавливать кюветы в кюветное отделение прибора всегда в строго определенное положение, чтобы избежать ошибок, связанных с различием в отражении и рассеянии света.
– Пользоваться одними и теми же кюветами при измерениях эталонных и испытуемого растворов.
– Накрывать кюветы крышками при работе с органическими растворителями во избежание их испарения.
–Не облучать растворы в течение длительного времени, так как при этом на внутренних стенках кювет появляются пузырьки газа, раствор иногда мутнеет, изменяется его светопоглощение. Для предотвращения подобных погрешностей нужно заново заполнить кюветы.
– Хранить кюветыв чистом виде, завернутыми в вату или мягкую бумагу, либо заполненными дистиллированной водой.
Приборы, реактивы: фотоэлектроколориметр КФК-5М с набором кювет, мерные колбы емкостью 100 мл, градуированные пипетки или бюретки.
Реактивы для определения Fe3+: соль железа (Ш) с содержанием 0,1 мг железа в 1 мл, 10 %-й растворсульфосалициловой кислоты,концентрированныйаммиак, 1 М раствор серной кислоты,сухой персульфат аммония, 50 %-й раствор роданида аммония, 1 М раствор соляной кислоты.
Реактивы для определения Cu2+: Соль меди с концентрацией 1 мг меди в 1 мл, 10 %-й раствораммиака.
Порядок выполнения работы
Определении концентрации Fe3+ методом градуировочного графика
1. Приготовить в мерных колбах серию эталонных растворов:
а) поместить 0, 1, 2, 3, 4, 5 мл раствора соли железа, 2 мл серной кислоты, 10 мл раствора сульфосалициловой кислоты, довести до метки водой и перемешать (раствор в начале серии служит контрольным опытом);
б) прилить 0, 1, 2, 3, 4, 5 мл раствора соли железа, 10 мл раствора сульфосалициловой кислоты, 5 мл раствора аммиака, разбавить водой до метки и размешать (первый раствор используется в качестве контрольного опыта);
в) разбавить исходный раствор соли железа в 20 раз и перемешать; в колбы ввести 0, 6, 10, 20, 30, 40, 50 мл этого раствора, 2 мл соляной кислоты, несколько крупинок персульфата аммония, 2 мл раствора роданида аммония, довести до метки водой и размешать (раствор без соли железа является контрольным опытом).
2. Подобрать, взяв раствор из середины серии, подходящие кюветы и светофильтр, при котором светопоглощение максимально.
3. Измерить оптическую плотность всех растворов относительно контрольного опыта.
4. Построить градуировочный график и установить верхнюю границу его линейности.
5. Разбавить анализируемый раствор в мерной колбе водой и перемешать. Взять в мерные колбы его аликвотные части (10 мл) и, проведя те же операции, как при изготовлении серии эталонных растворов, фотометрировать относительно контрольного опыта.
6. Найти по градуировочному графику концентрацию испытуемого раствора. Содержание железа в нем представить в виде 
 .
.
Определение концентрации Fe3+ методом сравнения
1. Рассчитать концентрацию исследуемого раствора, используя в качестве растворов сравнения эталонные растворы с оптической плотностью больше и меньше его светопоглощения.
2. Количество железа в нем представить в виде .
Определение концентрации Fe3+ методом добавок
Этот метод применяют при анализе растворов сложного состава, так как он позволяет автоматически учесть влияние «третьих» компонентов.
1. Взять в три мерные колбы одинаковые аликвотные части испытуемого раствора. В две колбы добавить разные, но точно известные количества определяемого элемента (с учетом соблюдения основного закона светопоглощения, установленного методом градуировочного графика). Прилить остальные компоненты так, как при изготовлении эталонных растворов, довести до метки водой и перемешать,
2. Измерить относительно контрольного опыта оптическую плотность анализируемого раствора и растворов с добавками, используя подходящие кюветы и длину волны максимального светопоглощения.
3. Концентрацию анализируемого раствора определить по формуле:
 (2.3)
(2.3)
где Ах – оптическая плотность анализируемого раствора; сх - концентрация определяемого компонента; сст - известное содержание определяемого компонента; которое добавляют в анализируемый раствор; Ах+ст – оптическая плотность раствора, содержащего определяемый компонент с добавкой.
4. Результаты представить в виде .
Определение концентрации Fe3+ по среднему значению изомолярного коэффициента погашения
1. Определить оптическую плотность стандартных растворов Аст1, Аст2, Аст3…
2. Для каждого раствора рассчитать молярный коэффициент погашения по формуле (2.1), а затем его усреднить.
2. Отобрать в мерные колбы аликвотные части испытуемого раствора, провести все необходимые операции и измерить оптическую плотность в подходящих кюветах при оптимальной длине волны.
3. Вычислить содержание железа (мг) в анализируемом растворе по формуле:
 (2.4)
(2.4)
представить результат в виде  .
.
Определение высоких концентраций растворов Cu2+ дифференциальным методом
Дифференциальные методы анализа применяют для определения больших количеств веществ, для устранения мешающего влияния посторонних примесей и исключения поглощения реактивов. Этот метод применяют еще и в тех случаях, когда из-за большой концентрации нарушается закон Бугера – Ламберта – Бера или когда значение оптической плотности выходит за границы шкалы прибора, а дальнейшее разбавление раствора нежелательно. Точность определения при использовании дифференциального метода повышается.
1. Приготовить в колбах эталонные растворы: ввести 5, 10, 15, 25, 30 мл раствора соли меди, 20 мл раствора аммиака, разбавить до метки водой и перемешать. Фотометрировать через 10 мин.
2. Подобрать подходящие кюветы и светофильтр, который обеспечивают максимальную оптическую плотность, проведя измерение оптической плотности раствора из середины серии по отношению к первому.
3. Фотометрировать все эталонные растворы относительно первого.
4. Построить градуировочный график.
5. Измерить оптическую плотность каждого эталонного раствора по отношению ко всем предыдущим растворам серии.
6. Вычислить по полученным данным фактор пересчета и усреднить его значение.

7. Провести с испытуемым раствором те же операции, которые выполнялись при изготовлении эталонных растворов.
8. Измерить его оптическую плотность относительно первого раствора и по графику найти концентрацию определяемого элемента.
9. Представить результаты в статистической форме.
Контрольные вопросы
1. Объясните, в чем заключается метод фотометрии.
2. Перечислите, какие методы фотометрии вы знаете.
3. Из каких этапов состоит фотометрическое исследование?
4. Какой закон применяется в фотометрическом методе анализа в дополнение к основному закону светопоглощения? В чем он заключается?
5. В чем заключается выбор оптимальной длины волны и толщины светопоглощающего слоя?
6. Дайте определение, что такое раствор сравнения и анализируемый раствор.
7. Перечислите правила использования кювет при фотометрии.
Лабораторная работа 3
КОЛИЧЕСТВЕННОЕ АТОМНО-АБСОРБЦИОННОЕ ОПРЕДЕЛЕНИЕ ПРЕДЛОЖЕННОГО МЕТАЛЛА В РАСТВОРЕ МЕТОДОМ КАЛИБРОВОЧНОГО ГРАФИКА
Цель работы:
1. Изучить устройство и порядок работы спектрометра SOLAAR М в режиме абсорбции в пламени.
2. Выполнить количественное определение предложенного металла в данном режиме с использованием калибровочного графика.