Гиперлипопротеинемия V типа
Этот тип гиперлипидемии во многом похож на I тип, но проявляется не только высокими хиломикронами, но и ЛПОНП.
23.Депонирование и мобилизация нейтрального жира в жировой ткани, регуляция процессов синтеза и мобилизации нейтрального жира.
Какой процесс будет преобладать в организме - синтез жиров (липогенез) или их распад (липолиз), зависит от поступления пищи и физической активности. В абсорбтивном состоянии под действием инсулина происходит липогенез, в постабсорбтивном состоянии - липолиз, активируемый глюкагоном. Адреналин, секреция которого увеличивается при физической активности, также стимулирует липолиз.
Регуляция синтеза жиров. В абсорбтивный период при увеличении соотношения инсулинглюкагон в печени активируется синтез жиров. В жировой ткани индуцируется синтез ЛП-липазы в адипоцитах и осуществляется её экспонирование на поверхность эндотелия; следовательно, в этот период увеличивается поступление жирных кислот в адипоциты. Одновременно инсулин активирует белки-переносчики глюкозы - ГЛЮТ-4. Поступление глюкозы в адипоциты и гликолиз также активируются. В результате образуются все необходимые компоненты для синтеза жиров: глицерол-3-фосфат и активные формы жирных кислот. В печени инсулин, действуя через различные механизмы, активирует ферменты путём дефосфорилирования и индуцирует их синтез. В результате увеличиваются активность и синтез ферментов, участвующих в превращении части глюкозы, поступающей с пищей, в жиры. Это - регуляторные ферменты гликолиза, пируватдегидрогеназный комплекс и ферменты, участвующие в синтезе жирных кислот из ацетил-КоА. Результат действия инсулина на обмен углеводов и жиров в печени - увеличение синтеза жиров и секреция их в кровь в составе ЛПОНП. ЛПОНП доставляют жиры в капилляры жировой ткани, где действие ЛП-липазы обеспечивает быстрое поступление жирных кислот в адипоциты, где они депонируются в составе триацилглицеринов. Запасание жиров в жировой ткани - основная форма депонирования источников энергии в организме человека. Запасы жиров в организме человека массой 70 кг составляют 10 кг, но у многих людей количество жиров может быть значительно больше. Жиры образуют в адипоцитах жировые вакуоли. Жировые вакуоли иногда заполняют значительную часть цитоплазмы. Скорость синтеза и мобилизации подкожного жира происходит неравномерно в разных частях организма, что связано с неодинаковым распределением рецепторов гормонов на адипоцитах.
24. Роль адреналина, глюкагона и инсулина в регуляции метаболизма нейтрального жира в жировой ткани.
Регуляция мобилизации жиров.Мобилизация депонированных жиров стимулируется глюкагоном и адреналином и, в меньшей степени, некоторыми другими гормонами (соматотроп-ным, кортизолом). В постабсорбтивный период и при голодании глюкагон, действуя на адипоциты через аденилатциклазную систему, активирует протеинкиназу А, которая фосфо-рилирует и, таким образом, активирует гормончувствительную липазу, что инициирует липо-лиз и выделение жирных кислот и глицерина в кровь. При физической активности увеличивается секреция адреналина, который действует через β-адренергические рецепторы адипоцитов, активирующие аденилатциклазную систему. В настоящее время обнаружено 3 типа β-рецепторов: β1, β2, β3, активация которых приводит к липолитическому действию. К наибольшему липолитическому действию приводит активация β3-рецепторов. Адреналин одновременно действует и на α2-рецепторы адипоцитов, связанные с ингибирующим G-белком, что инактивирует аденилатциклазную систему. Вероятно, действие адреналина двояко: при низких концентрациях в крови преобладает его антилиполитическое действие через α2-рецепторы, а при высокой - преобладает липолитическое действие через β-рецепторы.Для мышц, сердца, почек, печени при голодании или физической работе жирные кислоты становятся важным источником энергии. Печень перерабатывает часть жирных кислот в кетоновые тела, используемые мозгом, нервной тканью и некоторыми другими тканями как источники энергии. В результате мобилизации жиров концентрация жирных кислот в крови увеличивается приблизительно в 2 раза, однако абсолютная концентрация жирных кислот в крови невелика даже в этот период. Т1/2 жирных кислот в крови тоже очень мал (менее 5 мин), что означает существование быстрого потока жирных кислот из жировой ткани к другим органам. Когда постабсорбтивный период сменяется аборбтивным, инсулин активирует специфическую фосфатазу, которая дефосфорилирует гормончувствительную липазу, и распад жиров останавливается.

| Кровоток |
25. Физиологическая роль резервирования и мобилизации нейтрального жира и жировой ткани, нарушения процессов при ожирении.
Ожирением считают состояние, когда масса тела превышает 20% от "идеальной" для данного индивидуума. Образование адипоцитов происходит ещё во внутриутробном состоянии, начиная с последнего триместра беременности, и заканчивается в препубертатный период. После этого жировые клетки могут увеличиваться в размерах при ожирении или уменьшаться при похудании, но их количество не изменяется в течение жизни.
Первичное ожирение.Первичное ожирение характеризуется множеством гормональных и метаболических особенностей у лиц, страдающих этим заболеванием. В самом общем виде можно сказать, что первичное ожирение развивается в результате алиментарного дисбаланса - избыточной калорийности питания по сравнению с расходами энергии. Суточные потребности организма в энергии складываются из:
- основного обмена - энергии, необходимой для поддержания жизни; основной обмен измеряют по поглощению кислорода или выделению тепла человеком в состоянии покоя утром, после 12-часового перерыва в еде;
- энергии, необходимой для физической активности.
Затраты энергии, необходимые для физической активности, разделяют на 3 уровня:
· I - 30% энергии от основного обмена (у людей, ведущих сидячий образ жизни);
· II - 60-70% от энергии основного обмена (у людей, которые 2 ч в день имеют умеренную физическую нагрузку);
· III - 100% и более от энергии основного обмена (у людей, которые в течение нескольких часов в день занимаются тяжёлой физической работой).
В зависимости от интенсивности нагрузки и возраста суточная потребность в энергии колеблется у женщин от 2000 до 3000 ккал в день, а у мужчин - от 2300 до 4000 ккал. Количество потребляемой пищи определяется многими факторами, в том числе и химическими регуляторами чувства голода и насыщения. Эти чувства определяются концентрацией в крови глюкозы и гормонов, которые инициируют чувство насыщения: холецистокинина, нейротензина, бомбезина, лептина.
Причины первичного ожирения:
- генетические нарушенвся (до 80% случаев ожирения - результат генетических нарушений);
- состав и количество потребляемой пищи, метод питания в семье;
- уровень физической активности;
- психологические факторы.
Генетические факторы в развитии ожирения.Метаболические различия между тучными и худыми людьми до настоящего времени не могут быть определены однозначно. Существует несколько теорий, объясняющих эти различия:
· генетически детерминированная разница в функционировании "бесполезных" циклов (субстратных циклов, раздел 7). Эти циклы состоят из пары метаболитов, превращаемых друг в друга с помощью двух ферментов. Одна из этих реакций идёт с затратой АТФ. Например:

· если эти субстраты превращаются друг в друга с одинаковой скоростью, то происходит "бесполезный" расход АТФ и, соответственно, источников энергии, например жиров;
· у людей, склонных к ожирению, вероятно, имеется более прочное сопряжение дыхания и окислительного фосфорилирования, т.е. более эффективный метаболизм;
· возможно, разное соотношение аэробного и анаэробного гликолиза. Анаэробный гликолиз (как менее эффективный) "сжигает" гораздо больше глюкозы, в результате снижается её переработка в жиры;
· у отдельных ивдивидуумов имеется различие в активности Nа+/К+-АТФ:азы, работа которой требует до 30% энергии, потребляемой клетками.
У человека и животных имеется "ген ожирения" - obese gene (ob). Продуктом экспрессии этого гена служит белок лептин, состоящий из 167 аминокислот, который синтезируется и секретируется адипоцитами и взаимодействует с рецепторами гипоталамуса. В результате его действия снижается секреция нейропептида Y. Нейропептид Y стимулирует пищевое поведение, поиск и потребление пищи у животных. Другие пептиды, участвующие в регуляции чувства сытости, например холецистокинин, также влияют на секрецию нейропептида Y. Таким опосредованным путём лептин выступает регулятором жировой массы, необходимой для роста и репродукции. Уровень лептина у больных ожирением может быть различным. У 80% больных концентрация лептина в крови тучных людей больше в 4 раза, чем у людей с нормальной массой тела. В этих случаях имеется генетический дефект рецепторов лептина в гипоталамусе, поэтому, несмотря на продукцию лептина, центр голода в гипоталамусе продолжает секрецию нейропептида Y. 20% больных имеют изменения в первичной структуре лептина. К настоящему времени описаны 5 одиночных мутаций в гене лептина, которые приводят к развитию ожирения. У этих больных наблюдают повышение отложения жиров в жировой ткани, чрезмерное потребление пищи, низкую физическую активность и развитие сахарного диабета типа II. Патогенез ожирения при дефекте генаobможет быть следующим: низкий уровень лептина в крови служит сигналом недостаточного количества запаса жиров в организме; этот сигнал включает механизмы, приводящие к увеличению аппетита и в результате к увеличению массы тела. Следовательно, можно сделать вывод о том, что первичное ожирение - не просто следствие переедания, а результат действия многих факторов, т.е. ожирение - полигенное заболевание.
 Вторичное ожирение -ожирение, развивающееся в результате какого-либо основного заболевания, чаще всего эндокринного. Например, к развитию ожирения приводят гипотиреоз, синдром Иценко-Кушинга, гипогонадизм и многие другие заболевания
Вторичное ожирение -ожирение, развивающееся в результате какого-либо основного заболевания, чаще всего эндокринного. Например, к развитию ожирения приводят гипотиреоз, синдром Иценко-Кушинга, гипогонадизм и многие другие заболевания
Генетический дефект сфингомиелиназы - причина болезни Ниманна-Пика. Дети с таким дефектом погибают в раннем возрасте. Симптомы заболевания: увеличение печени и селезёнки (гепатоспленомегалия), в лизосомах которых накапливается сфингомиелин; умственная отсталость. Генетический дефект другого фермента (церамидазы) приводит к развитию болезни Фарбера, симптомами которой также являются гепато- и спленомегалия, а также поражение суставов (болезненность, отёчность).
Генетические дефекты лизосомных ферментов катаболизма гликосфинголипидов.В норме синтез и катаболизм гликосфинголипидов сбалансированы таким образом, что количество этих компонентов в мембранах постоянно. Если имеется генетический дефект какого-либо лизосомного фермента, участвующего в катаболизме гликосфинголипида, то в лизосомах накапливается не-деполимеризованный субстрат, так называемые "остаточные тельца", размеры лизосом увеличиваются, их мембрана может разрушаться, ферменты выходят в цитозоль, и функции клеток нарушаются. Генетические заболевания вследствие дефекта какого-либо из ферментов катаболизма гликосфинголипидов называют сфинголипидоза-ми, или лизосомными болезнями. Эти заболевания редки, но среди некоторых популяций людей их частота очень высока. Так, болезнь Гоше вследствие дефекта фермента β-глюкрзидазы у евреев встречается с частотой 166:100 000, болезнь Тея-Сакса (дефект фермента β-гексозаминидазы) - с частотой 33:100 000. Сфинголипидозы обычно приводят к смерти в раннем возрасте, так как происходит поражение клеток нервной ткани, где сконцентрированы гликосфинголипиды. Однако при болезнях Гоше и Фабри больные живут, относительно долго.
26. Обмен жирных кислот, b-окисление как специфический путь катаболизма жирных кислот, химизм, ферменты, энергетика.
β-Окисление - специфический путь катаболизма жирных кислот, при котором от карбоксильного конца жирной кислоты последовательно отделяется по 2 атома углерода в виде ацетил-КоА. Метаболический путь - β-окисление - назван так потому, что реакции окисления жирной кислоты происходят у β-углеродного атома. Реакции β-окисления и последующего окисления ацетил-КоА в ЦТК служат одним из основных источников энергии для синтеза АТФ по механизму окислительного фосфорилирования. β-Окисление жирных кислот происходит только в аэробных условиях.
Активация жирных кислот.Перед тем, как вступить в различные реакции, жирные кислоты должны быть активированы, т.е. связаны макроэргической связью с коферментом А:
RCOOH + HSKoA + АТФ → RCO ~ КоА + АМФ + PPi.
Реакцию катализирует фермент ацил-КоА син-тетаза. Выделившийся в ходе реакции пирофосфат гидролизуется ферментом пирофосфатазой: Н4Р2О7 + Н2О → 2 Н3РО4. Выделение энергии при гидролизе макроэргической связи пирофосфата смещает равновесие реакции вправо и обеспечивает полноту протекания реакции активации. Ацил-КоА синтетазынаходятся как в цитозоле, так и в матриксе митохондрий. Эти ферменты отличаются по специфичности к жирным кислотам с различной длиной углеводородной цепи. Жирные кислоты с короткой и средней длиной цепи (от 4 до 12 атомов углерода) могут проникать в матрикс митохондрий путём диффузии. Активация этих жирных кислот происходит в матриксе митохондрий. Жирные кислоты с длинной цепью, которые преобладают в организме человека (от 12 до 20 атомов углерода), активируются ацил-КоА синтетазами, расположенными на внешней мембране митохондрий.
Транспорт жирных кислот с длинной углеводородной цепью в митохондриях.β-Окисление жирных кислот, происходит в матриксе митохондрий, поэтому после активации жирные кислоты должны транспортироваться внутрь митохондрий. Жирные кислоты с длинной углеводородной цепью переносятся через плотную внутреннюю мембрану митохондрий с помощью карнитина. Карнитин поступает с пищей или синтезируется из незаменимых аминокислот лизина и метионина. В реакциях синтеза карнитина участвует витамин С (аскорбиновая кислота).В наружной мембране митохондрий находится фермент карнитинацилтрансфераза I (карнитин-пальмитоилтрансфераза I), катализирующий реакцию с образованием ацилкарнитина. Образовавшийся ацилкарнитин проходит через межмембранное пространство к наружной стороне внутренней мембраны и транспортируется с помощью карнитинацилкарнитинтранс-локазы на внутреннюю поверхность внутренней мембраны митохондрий, где фермент карнитинацилтрансфераза II катализирует перенос ацила на внутримитохондриальный КоА. Таким образом, ацил-КоА становится доступным для ферментов β-окисления. Свободный карнитин возвращается на цитозольную сторону внутренней мембраны митохондрий той же транслоказой. На внутренней поверхности внутренней мембраны находится фермент карнитинацил трансфераза II, катализирующий обратный перенос ацила с карнитина на внутримитохондриальный КоА. После этого ацил-КоА включается в реакции β-окисления. 
β-Окисление жирных кислот - специфический путь катаболизма жирных кислот, протекающий в матриксе митохондрий только в аэробных условиях и заканчивающийся образованием ацетил-КоА. Водород из реакций β-окисления поступает в ЦПЭ, а ацетил-КоА окисляется в цитратном цикле, также поставляющем водород для ЦПЭ. Поэтому β-окисление жирных кислот - важнейший метаболический путь, обеспечивающий синтез АТФ в дыхательной цепи.

β-Окисление начинается с дегидрирования ацил-КоА FAD-зависимой ацил-КоА дегидрогеназой с образованием двойной связи между α- и β-атомами углерода в продукте реакции - еноил-КоА. Восстановленный в этой реакции кофермент FADH2 передаёт атомы водорода в ЦПЭ на кофермент Q. В результате синтезируются 2 молекулы АТФ. В следующей реакции р-окисления по месту двойной связи присоединяется молекула воды таким образом, что ОН-группа находится у β-углеродного атома ацила, образуя β-гидроксиацил-КоА. Затем β-гидроксиацил-КоА окисляется NАD+-зависимой дегидрогеназой. Восстановленный NADH, окисляясь в ЦПЭ, обеспечивает энергией синтез 3 молекул АТФ. Образовавшийся β-кетоацил-КоА подвергается тиолитическому расщеплению ферментом тиолазой, так как по месту разрыва связи С-С через атом серы присоединяется молекула кофермента А. В результате этой последовательности из 4 реакций от ацил-КоА отделяется двухуглеродный остаток - ацетил-КоА. Жирная кислота, укороченная на 2 атома углерода, опять проходит реакции дегидрирования, гидратации, дегидрирования, отщепления ацетил-КоА. Эту последовательность реакций обычно называют "циклом β-окисления", имея в виду, что одни и те же реакции повторяются с радикалом жирной кислоты до тех пор, пока вся кислота не превратится в ацетильные остатки.
Продуктами каждого цикла β-окисления являются FADH2, NADH и ацетил-КоА.Хотя реакции в каждом "цикле" одни и те же, остаток кислоты, который входит в каждый последующий цикл, короче на 2 углеродных атома. В последнем цикле окисляется жирная кислота из 4 атомов углерода, поэтому образуются 2 молекулы ацетил-КоА, а не 1, как в предыдущих. Суммарное уравнение β-окисления, например пальмитоил-КоА может быть представлено таким образом:
С15Н31СО-КоА + 7 FAD + 7 NAD+ + 7 HSKoA → 8 СН3-СО-КоА + 7 FADH2 + 7 (NADH + H+).
Если рассчитывать выход АТФ при окислении пальмитиновой кислоты то из общей суммы молекул АТФ необходимо вычесть 2 молекулы, так как на активацию жирной кислоты тратится энергия 2 макроэргических связей. Следовательно, энергетический выход равен:

где n/2 – количество молекул Ацетил-КоА
n – количество С-атомов
12 – количество АТФ, получаемое при окислении Ацетил-КоА
n/2-1 – количество циклов β-окисления
5 – количество молекул АТФ, образовавшихся в цикле за счет 2 реакций дегидрирования
-1 – затрата одной АТФ
Судьба ацетил-КоА.
β-Окисление жирных кислот - специфический путь катаболизма жирных кислот, протекающий в матриксе митохондрий только в аэробных условиях и заканчивающийся образованием ацетил-КоА. Водород из реакций β-окисления поступает в ЦПЭ, а ацетил-КоА окисляется в цитратном цикле, также поставляющем водород для ЦПЭ. Поэтому β-окисление жирных кислот - важнейший метаболический путь, обеспечивающий синтез АТФ в дыхательной цепи.

β-Окисление начинается с дегидрирования ацил-КоА FAD-зависимой ацил-КоА дегидрогеназой с образованием двойной связи между α- и β-атомами углерода в продукте реакции - еноил-КоА. Восстановленный в этой реакции кофермент FADH2 передаёт атомы водорода в ЦПЭ на кофермент Q. В результате синтезируются 2 молекулы АТФ. В следующей реакции р-окисления по месту двойной связи присоединяется молекула воды таким образом, что ОН-группа находится у β-углеродного атома ацила, образуя β-гидроксиацил-КоА. Затем β-гидроксиацил-КоА окисляется NАD+-зависимой дегидрогеназой. Восстановленный NADH, окисляясь в ЦПЭ, обеспечивает энергией синтез 3 молекул АТФ. Образовавшийся β-кетоацил-КоА подвергается тиолитическому расщеплению ферментом тиолазой, так как по месту разрыва связи С-С через атом серы присоединяется молекула кофермента А. В результате этой последовательности из 4 реакций от ацил-КоА отделяется двухуглеродный остаток - ацетил-КоА. Жирная кислота, укороченная на 2 атома углерода, опять проходит реакции дегидрирования, гидратации, дегидрирования, отщепления ацетил-КоА. Эту последовательность реакций обычно называют "циклом β-окисления", имея в виду, что одни и те же реакции повторяются с радикалом жирной кислоты до тех пор, пока вся кислота не превратится в ацетильные остатки.
Продуктами каждого цикла β-окисления являются FADH2, NADH и ацетил-КоА.Хотя реакции в каждом "цикле" одни и те же, остаток кислоты, который входит в каждый последующий цикл, короче на 2 углеродных атома. В последнем цикле окисляется жирная кислота из 4 атомов углерода, поэтому образуются 2 молекулы ацетил-КоА, а не 1, как в предыдущих
28. Локализация ферментов b-окисления жирных кислот. Транспорт жирных кислот в митохондрии. Карнитин-ацилтрансфераза. (См. предыдущий вопрос)
29. Физиологическое значение процессов катаболизма жирных кислот.
30. Биосинтез пальмитиновой жирной кислоты, химизм, жирнокислотная синтетаза.
Установлено, что в цитоплазме пече-ночных клеток синтезируется пальмитиновая кислота (16 углеродных атомов), а в митохондриях этих клеток из уже синтезированной в цитоплазме клетки пальмитиновой кислоты или из жирных кислот экзогенного происхождения, т.е. поступающих из кишечника, образуются жирные кислоты, содержащие 18, 20 и 22 углеродных атома. Первой реакцией биосинтеза жирных кислот является карбоксилирование ацетил-КоА, для чего требуются бикарбонат, АТФ, ионы марганца. Катализирует эту реакцию фермент ацетил-КоА-кар-боксилаза. Фермент содержит в качестве простетической группы биотин. Реакция протекает в два этапа: I – карбоксилирование биотина с участием АТФ и II – перенос карбоксильной группы на ацетил-КоА, в результате чего образуется малонил-КоА. Малонил-КоА представляет собой первый специфический продукт биосинтеза жирных кислот. В присутствии соответствующей ферментной системы малонил-КоА быстро превращается в жирные кислоты. Последовательность реакций, происходящих при синтезе жирных кислот:

Далее цикл реакций повторяется.
31. Биосинтез жирных кислот с длинной цепью углеводных атомов (С18 и больше С-атомов).
Удлинение жирных кислот.В ЭР происходит удлинение пальмитиновой кислоты с участием малонил-КоА. Последовательность реакций сходна с той, что происходит при синтезе пальмитиновой кислоты, однако в данном случае жирные кислоты связаны не с синтазой жирных кислот, а с КоА. Ферменты, участвующие в элонгации, могут использовать в качестве субстратов не только пальмитиновую, но и другие жирные кислоты, поэтому в организме могут синтезироваться не только стеариновая кислота, но и жирные кислоты с большим числом атомов углерода.Основной продукт элонгации в печени - стеариновая кислота (С 18:0), однако в ткани мозга образуется большое количество жирных кислот с более длинной цепью - от С20 до С24, которые необходимы для образования сфинголипидов и гликолипидов. В нервной ткани происходит синтез и других жирных кислот - α-гидроксикислот. Оксидазы со смешанными функциями гидроксилируют С22 и С24 кислоты с образованием лигноцериновой и цереброновой кислот, обнаруживаемых только в липидах мозга.
Образование двойных связей в радикалах жирных кислот.Включение двойных связей в радикалы жирных кислот называется десатурацией. Основные жирные кислоты, образующиеся в организме человека в результате десатурации - пальмитоо-леиновая (С16:1Δ9) и олеиновая (С18:1Δ9). Образование двойных связей в радикалах жирных кислот происходит в ЭР в реакциях с участием молекулярного кислорода, NADH и цитохрома b5. Ферменты десатуразы жирных кислот, имеющиеся в организме человека, не могут образовывать двойные связи в радикалах жирных кислот дистальнее девятого атома углерода, т.е. между девятым и метильным атомами углерода. Поэтому жирные кислоты семейства ω-3 и ω-6 не синтезируются в организме, являются незаменимыми и обязательно должны поступать с пищей, так как выполняют важные регуляторные функции. Для образования двойной связи в радикале жирной кислоты требуется молекулярный кислород, NADH, цитохром b5 и FAD-зависимая редуктаза цитохрома b5. Атомы водорода, отщепляемые от насыщенной кислоты, выделяются в виде воды. Один атом молекулярного кислорода включается в молекулу воды, а другой также восстанавливается до воды с участием электронов NADH, которые передаются через FADH2 и цитохром b
32. Биосинтез ненасыщенных кислот. Полиненасыщенные жирные кислоты
Выделяют два семейства полиненасыщенных жирных кислот: омега-3 и омега-6. Жиры от каждой из этих семейств являются существенными, поскольку организм может преобразовать одну омегу-3 в другую омегу-3, например, но не может создать омегу-3 на пустом месте. Вместе весь комплекс полиненасыщенных жиров называется витамин F. В эту группу входит комплекс полиненасыщенных жирных кислот, которые принимают значительное участие в биологических процессах:
· линолевая кислота (омега-6)
· линоленовая кислота (омега-3)
· арахидоновая кислота (омега-6)
· эйкозапентаеновая кислота (омега-3)
· докозагексаеновая кислота (омега-3)
Полиненасыщенные жирные кислоты препятствуют развитию атеросклероза и снижают уровень триглицеридов, липопротеидов низкой плотности в крови, холестерина и его отложение на стенках артерий. Линолевая кислота синтезирует простагландины, которые способны снижать давление. Витамин F влияет на агрегационную активность тромбоцитов, а также уменьшает содержание фибриногена в крови, то есть способствует разжижению крови, оказывая антитромботическое действие на сердечно-сосудистую систему. Полиненасыщенные жирные кислоты обладают кардиопротекторным и антиаритмическим действием. Полиненасыщенные жирные кислоты участвуют в биосинтезе эйкозаноидов – гормонов местного действия.
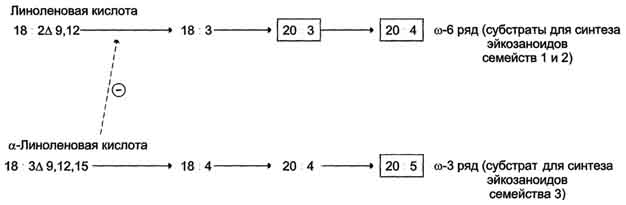
Главный субстрат для синтеза эйкозаноидов у человека - арахидоновая кислота (20:4, ω-6), так как её содержание в организме человека значительно больше остальных полиеновых кислот-предшественников эйкозаноидов. В меньшем количестве для синтеза эйкозаноидов используются эйкозапентаеновая (20:5, ω-3) и эйкозатриеновая (20:3, ω-6) жирные кислоты. Полиеновые кислоты с 20 атомами углерода поступают в организм человека с пищей или образуются из незаменимых (эссенциальных) жирных кислот с 18 атомами углерода, также поступающими с пищей. Полиеновые жирные кислоты, которые могут служить субстратами для синтеза эйкозаноидов, входят в состав глицерофосфолипидов. мембран. Под действием ассоциированной с мембраной фосфолипазы А2 жирная кислота отщепляется от глицерофосфолипида и используется для синтеза эйкозаноидов.
33. Биосинтез и использование ацетоуксусной кислоты, физиологическое значение процессов.
При голодании, длительной физической работе и в случаях, когда клетки не получают достаточного количества глюкозы, жирные кислотыиспользуются многими тканями как основной источник энергии. В отличие от других тканей мозг и другие отделы нервной ткани практически не используют жирные кислоты в качестве источника энергии. В печени часть жирных кислот превращается в кетоновые тела, которые окисляются мозгом, нервной тканью, мышцами, обеспечивая достаточное количество энергии для синтеза АТФ и уменьшая потребление глюкозы. К кетоновымтелам относят β-гидроксибутират, ацетоацетат и ацетон. Первые две молекулы могут окисляться в тканях, обеспечивая синтез АТФ. Ацетон образуется только при высоких концентрациях кетоновых тел в крови и, выделяясь с мочой, выдыхаемым воздухом и потом, позволяет организму избавляться от избытка кетоновых тел.
Синтез кетоновых тел в печени.При низком соотношении инсулин/глюкагон в крови в жировой ткани активируется распад жиров. Жирные кислоты поступают в печень в большем количестве, чем в норме, поэтому увеличивается скорость β-окисления. Скорость реакций ЦТК в этих условиях снижена, так как оксалоацетат используется для глюконеогенеза. В результате скорость образования ацетил-КоА превышает способность ЦТК окислять его. Ацетил-КоА накапливается в митохондриях печени и используется для синтеза кетоновых тел. Синтез кетоновых тел происходит только в митохондриях печени.

Синтез кетоновых тел начинается с взаимодействия двух молекул ацетил-КоА, которые под действием фермента тиолазы образуют ацетоацетил-КоА. С ацетоацетил-КоА взаимодействует третья молекула ацетил-КоА, образуя 3-гидрокси-3-метилглутарил-КоА (ГМГ-КоА). Эту реакцию катализирует фермент ГМГ-КоА-синтаза. Далее ГМГ-КоА-лиаза катализирует расщепление ГМГ-КоА на свободный ацетоацетат и ацетил-КоА. Ацетоацетат может выделяться в кровь или превращаться в печени в другое кетоновое тело - β-гидроксибутират путём восстановления. В клетках печени при активном β-окислении создаётся высокая концентрация NADH. Это способствует превращению большей части ацетоацетата в β-гидроксибутират, поэтому основное кетоновое тело в крови - именно β-гидроксибутират. При голодании для многих тканей жирные кислоты и кетоновые тела становятся основными топливными молекулами. Глюкоза используется в первую очередь нервной тканью и эритроцитами. При высокой концентрации ацетоацетата часть его неферментативно декарбоксилируется, превращаясь в ацетон. Ацетон не утилизируется тканями, но выделяется с выдыхаемым воздухом и мочой. Таким путём организм удаляет избыточное количество кетоновых тел, которые не успевают окисляться, но, являясь водорастворимыми кислотами, вызывают ацидоз. При длительном голодании кетоновые тела становятся основным источником энергии для скелетных мышц, сердца и почек. Таким образом глюкоза сохраняется для окисления в мозге и эритроцитах. Уже через 2-3 дня после начала голодания концентрация кетоновых тел в крови достаточна для того, чтобы они проходили в клетки мозга и окислялись, снижая его потребности в глюкозе. β-Гидроксибутират , попадая в клетки, дегидрируется NAD-зависимой дегидрогеназой и превращается в ацетоацетат. Ацетоацетат активируется, взаимодействуя с сук-цинил-КоА - донором КоА:
Ацетоацетат + Сукцинил-КоА → Ацетоацетил- КоА + Сукцинат.
Реакцию катализирует сукцинил-КоА-ацето-ацетат-КоА-трансфераза. Этот фермент не синтезируется в печени, поэтому печень не использует кетоновые тела как источники энергии, а производит их "на экспорт". Кетоновые тела - хорошие топливные молекулы; окисление одной молекулы β-гидроксибутирата до СО2 и Н2О обеспечивает синтез 27 молекул АТФ. Эквивалент одной макроэргической связи АТФ (в молекуле сукцинил-КоА) используется на активацию ацетоацетата, поэтому суммарный выход АТФ при окислении одной молекулы β-гидроксибутирата - 26 молекул.
34. Обмен стероидов. Холестерин как предшественник других стероидов. Биосинтез холестерина.
Холестерол - стероид, характерный только для животных организмов. Он синтезируется во многих тканях человека, но основное место синтеза - печень. В печени синтезируется более 50% холестерола, в тонком кишечнике - 15- 20%, остальной холестерол синтезируется в коже, коре надпочечников, половых железах. В сутки в организме синтезируется около 1 г холестерола; с пищей поступает 300-500 мг Холестерол выполняет много функций: входит в состав всех мембран клеток и влияет на их свойства, служит исходным субстратом в синтезе жёлчных кислот и стероидных гормонов. Предшественники в метаболическом пути синтеза холестерола превращаются также в убихинон - компонент дыхательной цепи и долихол, участвующий в синтезе гликопротеинов. Холестерол за счёт своей гидроксильной группы может образовывать эфиры с жирными кислотами. Этерифицированный холестерол преобладает в крови и запасается в небольших количествах в некоторых типах клеток, использующих его как субстрат для синтеза других веществ. Холестерол и его эфиры - гидрофобные молекулы, поэтому они транспортируются кровью только в составе разных типов ЛП. Обмен холестерола чрезвычайно сложен - только для его синтеза необходимо осуществление около 100 последовательных реакций. Всего в обмене холестерола участвует около 300 разных белков. Нарушения обмена холестерола приводят к одному из наиболее распространённых заболеваний - атеросклерозу. Смертность от последствий атеросклероза (инфаркт миокарда, инсульт) лидирует в общей структуре смертности населения. Атеросклероз - "полигенное заболевание", т.е. в его развитии участвуют многие факторы, важнейшие из которых наследственные. Накопление холестерола в организме приводит к развитию и другого распространённого заболевания - желчнокаменной болезни.
Синтез холестерола и егорегуляцияРеакции синтеза холестерола происходят в цитозоле клеток. Это один из самых длинных метаболических путей в организме человека.
Образование мевалоната
Сложный путь синтеза холестерола можно разделить на 3 этапа

Первый этап заканчивается образованием мевалоната (мевалоновой кислоты). Две молекулы ацетил-КоА конденсируются ферментом тиолазой с образованием ацетоацетил-КоА. Фермент щдроксиметилглутарил-КоА-синтаза присоединяет третий ацетильный остаток с образованием ГМГ-КоА (3-гидрокси-3-метилглутарил-КоА). Эта последовательность реакций сходна с начальными стадиями синтеза кетоновых тел:

Далее реакция, катализируемая ГМГ-КоА-редуктазой, является регуляторной в метаболическом пути синтеза холестерола. В этой реакции происходит восстановление ГМГ-КоА до мевалоната с использованием 2 молекул NADPH. Фермент ГМГ-КоА-редуктаза - гликопротеин, пронизывающий мембрану ЭР, активный центр которого выступает в цитозоль.
Образование скваленаНа втором этапе синтеза мевалонат превращается в пятиуглеродную изопреноидную структуру, содержащую пирофосфат - изопентенилпирофосфат. Продукт конденсации 2 изопреновых единиц - геранилпирофосфат. Присоединение ещё 1 изопреновой единицы приводит к образованию фарнезилпирофосфата - соединения, состоящего из 15 углеродных атомов. Две молекулы фарнезилпирофосфата конденсируются с образованием сквалена - углеводорода линейной структуры, состоящего из 30 углеродных атомов.
Образование холестерола.На третьем этапе синтеза холестерола сквален через стадию образования эпоксида ферментом циклазой превращается в молекулу ланостерола, содержащую 4 конденсированных цикла и 30 атомов углерода. Далее происходит 20 последовательных реакций, превращающих ланостерол в холестерол. На последних этапах синтеза от ланостерола отделяется 3 атома углерода, поэтому холестерол содержит 27 углеродных атомов.У холестерола имеется насыщенная разветвлённая боковая цепь из 8 углеродных атомов в положении 17, двойная связь в кольце В между атомами углерода в положениях 5 и 6, а также гидроксильная группа в положении 3.В организме человека изопентенилпирофосфат также служит предшественником убихинона (KoQ) и долихола, участвующего в синтезе гликопротеинов.
Этерификация холестерола.В некоторых тканях гидроксильная группа холестерола этерифицируется с образованием более гидрофобных молекул - эфиров холестерола. Реакция катализируется внутриклеточным ферментом АХАТ (ацилКоА:холестеролаиилтрансферазой). Реакция этерификации происходит также в крови в ЛПВП, где находится фермент ЛХАТ (лецитин:холестеролацилтрансфераза). Эфиры холестерола - форма, в которой они депонируются в клетках или транспортируются кровью. В крови около 75% холестерола находится в виде эфиров