Тератогенное действие лекарств
Лекция № 4
Врожденные и наследственные заболевания
I. Понятие врожденных и наследственных заболеваний. Их отличия. Генокопии и фенокопии. Мультифакториальные болезни.
Врожденные заболевания – заболевания, возникающие внутриутробно (пренатально), в период родов (интернатально) и существующие к моменту рождения.
Врожденные заболевания могут быть наследственными и ненаследственными, причем более распространены ненаследственные врожденные заболевания.
Наследственные заболевания – обязательно сопровождаются поражением генетического аппарата, передаются по наследству.
Большая часть наследственных заболеваний проявляется сразу после рождения и является врожденной патологией.
Таким образом, не все врожденные заболевания наследственные и есть часть наследственных болезней, не являющихся врожденными.
Фенокопия – ненаследственное изменение фенотипа организма, вызванное факторами окружающей среды и копирующее проявление какого-либо известного наследственного изменения (заболевания). Причиной фенокопии служит нарушение обычного хода индивидуального развития без изменения генотипа.
Генокопия - возникновение внешне сходных фенотипических признаков (заболеваний) под воздействием генов, расположенных в различных участках хромосомы или в различных хромосомах, т.е. заболевание предопределяется разными генами. Например, слепота может быть связана с генетическим поражением и сетчатки и хрусталика, которые контролируются различными генами. Существует несколько генокопий синдрома Дауна.
Врожденная патология, вызванная нарушениями развития плода, наблюдается у приблизительно 2% новорожденных и является наиболее частой причиной неонатальной смертности и заболеваемости. При большинстве аномалий не обнаруживается никаких хромосомных нарушений, и они не являются наследственными.
Среди наследственных болезней выделяют болезни с наследственной предрасположенностью (мультифакториальные). Наследственная предрасположенность подразумевает, что болезнь не детерминируется жестко генетическим аппаратом, но по наследству передаются некие свойства и особенности организма, его органов и систем, которые предрасполагают к возникновению определенных болезней (атеросклероз, гипертоническая болезнь, сахарный диабет, опухоли и др.). В основе мультифакториальных болезней лежит полигенное наследование, когда многие пары генов суммируют свое влияние (аддитивное действие)
Основные понятия генетики
Кариотип - набор хромосом. Нормальные клетки человека имеют 46 хромосом: 22 пары аутосом (соматические)и две половые хромосомы, по одной хромосоме в каждой паре человек получает от каждого родителя (46 ХY - мужчина, 46 ХХ – женщина).Половые хромосомы- это пара Х хромосом у женщин и Х и Y хромосомы у мужчин.
Диплоидный набор хромосом в соматических клетках (образуется при митозе).
Гаплоидный набор хромосом в половых клетках - гаметах (образуется при мейозе).
Ген - участок ДНК отвечает за синтез белка. ДНК (азотистые основания (А, Г, Ц, Т), дезоксирибоза и остаток фосфорной кислоты) представляет собой двойную спираль с комплементарностью. Триплет (кодон) – кодирует синтез 1 аминокислоты.
Генотип (кариотип) – совокупностей особенностей хромосомного набора индивидуума. Фенотип – совокупность проявившихся признаков организма при его взаимодействии со средой.
Аллели – контролируют альтернативные варианты одного и того же признака. Организм который имеет два одинаковых аллеля одного гена (ВВ или вв) – гомозиготен, разные аллели (Вв) – гетерозиготен.
Наследственная изменчивость бывает двух видов. Комбинативная - перетасовка неизменных генов. Мутационная - изменение генотипа, со скочкообразным изменением признаков.
Плейотропизм гена - 1 ген отвечает за разные признаки (синдром Марфана у Паганини - арахнодактилия, катаракта, аномалии скелета, пороки сердца).
Пенетрантность - проявляемость, вероятность проявления признака, закодированных геном (частота, с которой аномальный ген проявляется себя клинически в виде болезни).
Экспрессивность - степень проявления признака (степень выраженности клинических проявлений аномального гена у различных индивидуумов).
Сибсы – дети одной родительской пары.
Пробанд – лицо, через которое регистрируется вся семья (обычно впервые обратившийся из описываемой семьи).
Не весь генетический материал локализован в хромосомах. Существует митохондриальный геном (мтДНК) – небольшая кольцевая молекула, содержит 13 генов, кодирует синтез цитохром-оксидаз, АТФаз и др.
II. Тератогены. Классификация врожденных заболеваний в зависимости от срока возникновения. Тератогенное действие лекарств.
Тератогенные факторы (или тератогены) - факторы, вызывающие пороки развития (от греч. teratos - уродство).
Врожденные заболевания подразделяются в зависимости от срока возникновения.
1) период прогенеза соответствует созреванию гамет (яйцеклеток и сперматозоидов) до оплодотворения (в этот период возможно возникновение патологии гамет – гаметопатии);
2) период киматогенеза (от греч. kyema - зародыш) соответствует периоду от оплодотворения до родов. С периодом киматогенеза совпадает период киматопатии. В нем различают три периода:
· бластогенез - период от оплодотворения до 15 дня беременности. В этот период идет дробление яйца, заканчивается образованием эмбриобласта и трофобласта (в этот период возможно возникновение бластопатии);
· эмбриогенез - период с 16 дня до 75 дня беременности, идет основной органогенез и образуется амнион и хорион (в этот период возможно возникновение эмбриоопатии);
· фетогенез - период с 76 дня по 280 день беременности, происходит дифференцировка и созревание тканей плода, образование плаценты, а также рождение плода (в этот период возможно возникновение фетопатии). Фетогенез в свою очередь делится на
· ранний фетальный период (76-180 день беременности) - возможно возникновение болезней раннего фетогенеза;
· поздний фетальный период (181-280 день беременности) - возможно возникновение болезней позднего фетогенеза.
Таблица 4.1.
Критические периоды эмбриогенеза человека, в неделях (Мур, 1973)
| Деление зиготы, имплантация | Эмбриональный период | Плодный период | Роды | ||||||||
| 20-36 | |||||||||||
| Зародыш обычно погибает в присутствии тератогенов | Центральная нервная система | ||||||||||
| Сердце | |||||||||||
| Верхние конечности | |||||||||||
| Глаза | |||||||||||
| Нижние конечности | |||||||||||
| Зубы | |||||||||||
| Твердое небо | |||||||||||
| Наружные половые органы | |||||||||||
| Уши | |||||||||||
Наиболее изученными причинами киматопатий являются следующие:
Ионизирующее излучение:кроме прямого действия воздействия на ДНК и генетический аппарат клетки, ионизирующая радиация обладает прямым токсическим эффектом на клетки развивающегося плода и является причиной многих врожденных аномалий в результате воздействия ее в период беременности;
Тератогенные вирусные инфекции: вирус краснухи является наиболее сильным тератогенным вирусом и служит причиной большого количества врожденных дефектов. Трансплацентарное инфицирование плода вирусом во время первого триместра беременности, приводит к развитию большого количества врожденных аномалий. Вирус краснухи нарушает синтез белков в тканевых культурах. Синдром краснухи объединяет триаду врожденных аномалий: пороки сердца, глухоту и катаракту. Также были описаны случаи микроцефалии, умственной отсталости и микроофтальмии. Тератогенный эффект других вирусных инфекций дискутируется. Имеются сообщения о тератогенном действии вирусов гриппа, паротита (свинки) и ветряной оспы.
Тератогенное действие лекарств
Нельзя однозначно сказать, в каком периоде внутриутробного развития фармакотерапия может привести к более тяжелым последствиям. Наиболее масштабными по последствиям обычно являются повреждения, полученные в периоды бласто- и эмбриогенеза, хотя в некоторых случаях даже нарушения в периоде фетогенеза могут быть несовместимыми с жизнью. Правда, в периоде фетогенеза для защиты плода, в том числе от лекарств - ксенобиотиков возникает дополнительный защитный барьер – плацента.
Во время беременности необходимо по возможности исключить прием лекарств любого типа, за исключением случаев, когда это необходимо для спасения жизни матери или плода. Нет лекарств, которые могут быть признаны полностью безопасными, особенно во время ранних сроков беременности. Несмотря на то, что все применяемые лекарства, проходят испытания на беременных животных, безопасность их для людей может быть установлена только после моголетнего их использования, как это случилось в США при использовании талидомида и диэтилстильбэстрола.
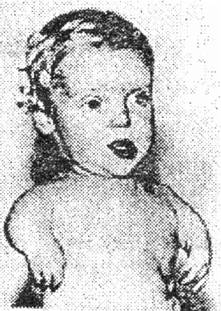
Рис. 4.1. Тератогенный эффект талидомида. Фокомелия у новорожденного младенца, мать которого принимала талидомид в течение двух первых месяцев беременности.
Талидомид является слабым седативным препаратом, который широко использовался в Европе в 60-х годах, пока не появились эпидемиологические доказательства того, что он является причиной определенных нарушений развития плода (фокомелии) при использовании его во время беременности. В результате нарушения развития конечностей ноги и руки ребенка напоминают ласты моржа - короткие культи конечностей, близко расположенные к туловищу.
Диэтилстильбэстрол является синтетическим эстрогеном, который широко использовался в 50-х годах для лечения угрожающего аборта. У дочерей женщин, принимавших диэтилстильбэстрол, часто развивались эпителиальные аномалии влагалища, включая увеличение количества слизистых желез (вагинальный аденоз), в более тяжелых случаях - светлоклеточную аденокарциному.
Главным условием формирования будущего организма является, прежде всего, нормальная закладка и оптимальное протекание биосинтетических (прежде всего биосинтеза белка) процессов, размножения клеток; а в более позднем внутриутробном периоде - периоде фетогенеза - и нормальное функционирование сформировавшихся органов.
Для биосинтетических процессов нужны строительные материалы (прежде всего – аминокислоты) и достаточное количество энергии АТФ, для получения которой, в свою очередь, требуются субстраты окисления, кислород и ферменты (включая витамины и микроэлементы).
Опасность представляют все препараты, прямо блокирующие биосинтез белка (например, большинство антибиотиков), либо косвенно препятствующие биосинтезу (например, глюкокортикоидные препараты, не только прямо блокирующие биосинтез многих белков, но также переводящие глюкогенные аминокислоты в глюкозу в ходе глюконеогенеза и лишающие процесс биосинтеза главного строительного материала).
Крайне опасны препараты, ограничивающие митотическую активность клетки (все группы противоопухолевых препаратов, так называемые цитостатики).
Биосинтетичесие процессы будут блокированы и в случае дефицита АТФ - при назначении препаратов, блокирующих на разных стадиях тканевое дыхание, включая разобщители окисления и фосфорилирования (многие представители упомянутых выше химиотерапевтических средств, т.е. средств, истребляющих чужеродные субклеточные (вирусы), клеточные (бактерии, простейшие, опухолевые клетки), многоклеточные (глисты), аналоги гормонов щитовидной железы или препараты, наоборот, нарушающие синтез этих гормонов).
Опасны препараты, вмешивающиеся в синтез и реализацию эффектов не только многих классических, но и так называемых тканевых гормонов - простагландинов, которые косвенно, через механизмы активации ферментов, участвуют в процессах биосинтеза белка (нестероидные противовоспалительные средства), антикальциевые препараты, вещества, блокирующие действия нейромедиаторов (блокаторы адрено-, холино- опиатных и гистаминовых рецепторов).
Косвенно нарушать снабжение плода аминокислотами, витаминами и глюкозой способны средства, понижающие аппетит (анорексики), принимаемые иногда женщинами для похудения, либо препараты, снижающие всасывание вышеперечисленных средств в желудочно-кишечном тракте матери (сорбенты, "кишечная" группа антибиотиков, средства, усиливающие перистальтику, либо ослабляющие секрецию пищеварительных желез или желчи печенью).
Возникновение патологии плода при косвенном влиянии (на организм матери) возможно, также вследствие назначения всех лекарственных препаратов, так или иначе вызывающих гипоксию (нарушение биоокисления) - угнетающих дыхательный центр (опиаты, "стволовые" снотворные), снижающих кровообращение плаценты - сосудорасширяющих средств и средств, угнетающих работу сердца из-за падения системного артериального давления; либо наоборот, сосудосуживающих, вызывающих ишемию плаценты при сохранном либо даже повышенном системном АД.
Опасны препараты, нарушающие снабжение плода глюкозой (гипогликемические) или блокирующие перенос гемоглобином кислорода (метгемоглобинобразователи типа жаропонижающих - производных анилина).
Период бласто- и эмбриогенеза, как уже говорилось, является наиболее уязвимым ещё и потому, что отсутствует плацентарный барьер, поэтому в будущий организм попадают все вещества, находящиеся в крови матери, независимо от их гидро- и липофильности. В период фетогенеза из тех же соображений более опасны будут липофильные лекарственные препараты. Вероятность тератогенного действия лекарственных веществ может повыситься при совместном применении их с гепатотоксичными препаратами, поскольку будут нарушаться детоксикационные механизмы, например, так называемые защитные синтезы и микросомальное окисление, направленные на придание молекуле гидрофильности, что должно было хотя бы частично ограничивать проникновение этих лекарств через плаценту.
III. Механизм формирования патологии у плода при курении беременной женщины.
Никотин, монооксид углерода (СО) и другие токсичные компоненты табачного дыма легко проникают в плаценту, снижая поступление кислорода к плоду, а также влияя на структуру и функционирование пуповины и плаценты. Никотин оказывает прямое воздействие на сердечный ритм. Эффекты никотина в данном случае обусловлены его возбуждающим действием на Н-холинорецепторами клеток мозгового вещества надпочечников, что стимулирует выброс последними катехоламинов. Адреналин и норадреналин, возбуждая b-адренорецепторы миокарда, способствуют увеличению силы и частоты сердечных сокращений (отсюда – аритмии), а стимуляция a-адренорецепторов сосудов пуповины и плаценты, а также внутренних органов плода ограничивает приток богатой кислородом крови к тканям плода. Кроме того, возбуждение b-адренорецепторов жировой ткани стимулирует ее распад, следовательно, возможно развитие гипотрофии плода.
Монооксид углерода (СО) обладает очень высоким сродством к гемоглобину (в 300 большим, чем О2), поэтому способен необратимо связываться с гемоглобином, образуя лишенный способности транспортировать и отдавать кислород комплекс – карбоксигемоглобин (HbCO). Следствием чего является гемическая гипоксия.
Целый ряд проникающих через плаценту компонентов табачного дыма являются известными канцерогенами (например, 5-бензпирен).
У курящих беременных:
- значительно чаще происходят самопроизвольные аборты и осложнения беременности и родов.
- более высокий риск эктопической (трубной) беременности.
- большая вероятность преждевременных родов.
- на 20-50% более высокий риск смерти плода и младенца.
- материнское курение может предрасположить ребенка к респираторным заболеваниям. Исследования дали основания считать, что курение любого из родителей в течение беременности сопровождается более высокой вероятностью возникновения всех видов детского рака, вместе взятых.
- ухудшение поведенческих, интеллектуальных и физических характеристик будущего ребенка.
· новорожденные курящих матерей имеют "симптомы отмены никотина".
IV. Механизм формирования патологии у плода при алкоголизме беременной женщины.
Алкоголь проходит через плаценту и попадает в организм плода. Вреден и сам спирт (этанол), и продукты его распада – например, ацетальдегид.
Спирт может вызывать спазм сосудов плаценты и пуповины, что приводит к кислородному голоданию плода.
Ацетальдегид:
· снижает уровень цинка в плодовых клетках, что нарушает их рост и развитие;
· способен вызывать мутации в ДНК зародышевых клеток, что может привести к грубым порокам развития органов и тканей плода.
Кроме того, алкоголь обусловливает дефицит витаминов, нарушает обмен веществ (гормонов, простагландинов и т.д.). В итоге страдают многие органы и системы плода, но более всего - центральная нервная система (в том числе интеллект и поведенческая сфера).
Употребление алкоголя женщиной во время беременности значительно повышает риск невынашивания, рождения маловесных детей, мертворождению, а в наиболее тяжелых случаях - развитию алкогольного синдрома плода (АСП) или синдром алкогольной фетопатии. Так называют болезнь плода, обусловленную внутриутробным алкогольным повреждением.
Для этого синдрома характерны врожденные аномалии развития сердца, наружных половых органов, нарушение функции центральной нервной системы, низкая масса тела при рождении, нарушения строения позвоночника, включая spina bifida, отставание ребенка в физическом и умственном развитии.
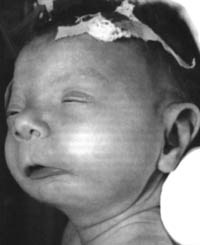
Рис. 4.2. Ребенок с синдромом алкогольной фетопатии. Заметны характерные черты лица: маленькая голова, в особенности лицо, низкий лоб, низко посаженные уши, узкие глаза, недоразвитие подбородка, специфическая складка век, большой рот с тонкими губами, выпуклой верхней губой и узкой красной каймой ("рот рыбы").
Последствия алкогольного поражения плоданеобратимы и практически не поддаются лечению.
Характер последствий алкогольного воздействия на плод зависит от очень многих причин. Безусловно, важнейшую роль играют количество спиртных напитков и частота их употребления. Ежедневный прием беременной 30 граммов спирта (или других алкогольных напитков в пересчете на спирт), сопровождается высоким риском синдрома алкогольной фетопатии у будущего ребенка. Однако, нередко этот синдром встречается у детей, матери которых употребляли 3-5 граммов спирта ежедневно! Единой безопасной для всех дозы не существует, т.к. вредность алкоголя для плода зависит также от особенностей женского организма по переработке спирта и реакции на алкоголь, состояния органов и систем женщины на момент беременности, характера диеты, сопутствующих вредных привычек, генотипа плода, срока беременности и многих других моментов.
V. Методы изучения наследственных болезней.
Клинико-генеалогический метод заключается в составлении родословной записи с последующим анализом проявления признака, характерного для конкретной наследственной болезни на протяжении возможно большего числа поколений родственников пациента.
Признаками наследственных болезней, установленных с помощью «родословных», являются:
1) обнаружение болезни «по вертикали»: из поколения в поколение беспрерывно (при доминантном типе наследования) или с некоторыми перерывами (при рецессивном типе наследования);
2) менделевские соотношения между числом больных и здоровых сибсов (3:1; 1:1; 1:0);
3) большая частота заболевания среди родственников, чем среди неродственников.
Близнецовый метод состоит в сопоставлении внутрипарной конкордантности (идентичности) одно- и двуяйцевых близнецов, живущих в разных и в одинаковых условиях, по анализируемому патологическому признаку.
В среднем на каждые 100 одноплодных родов приходятся одни близнецовые (многоплодные); при этом однояйцевые близнецы рождаются реже, чем двуяйцевые, примерно в 3-4 раза.
О наследственной природе патологии свидетельствует высокая конкордантность по анализируемому признаку однояйцевых близнецов, живущих в разных условиях, и, наоборот, низкая конкордантность двуяйцевых близнецов, особенно живущих в одинаковых условиях. Напротив, высокая конкордантность по какому-либо патологическому признаку одно- и двуяйцевых близнецов, живущих в одинаковой среде, явно говорит против наследственного происхождения данной патологии и, наоборот, подтверждает решающее значение в ее развитии экзогенных (внешних) факторов.
Популяционно-статистический метод заключается в составлении родословных среди большой группы населения, в пределах области или целой страны, в исследовании генетических изолятов. Изолят - это группа людей, от 500 человек до нескольких тысяч, живущая изолированно от всего остального населения страны. Генетический изолят характеризуется тем, что браки заключаются только в его пределах, с высокой частотой эндогамных браков. Это ведет в конце концов к генной изоляции от остального народа страны. В результате происходит передача аномальных рецессивных генов из гетерозиготных в гомозиготные пары, что сопровождается увеличением числа наследственных болезней.
Цитологический метод -установление генетического пола при исследовании клеток на наличие телец Барра. Когда в клетке присутствует две Х хромосомы (как у нормальной женщины), одна из них (тельце Барра) инактивируется и конденсируется на ядерной мембране. Отсутствие тельца Барра свидетельствует о наличии только одной Х хромосомы (у нормального мужчины (XY) и при синдроме Шершевского-Тернера (ХО)). Тельца Барра наиболее легко определяются в мазках многослойного эпителия, которые получают путем соскабливания буккальной слизистой оболочки.
Биохимический и иммунологический методы заключаются в исследовании биохимических признаков, заведомо специфичных для определенных наследственных болезней. Так, например, для диагностики фенилпировиноградной олигофрении в моче определяют фенилпировиноградную кислоту; для диагностики серповидно-клеточной анемии (S-гемоглобиноза) исследуют наличие в крови S-гемоглобина; для выявления иммунодефицитных состояний определяют содержание различных антител и популяций лимфоцитов.
Дерматоглифический метод – выявление наследственных болезней по рисунку ладоней.
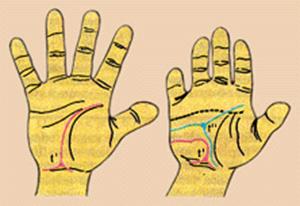
Рис. 4.3. Схематическое изображение ладони здорового ребенка (слева) и ребенка того же возраста с болезнью Дауна.
Цитогенетический метод состоит в микроскопическом исследовании структуры и числа хромосом клеток (лейкоцитов, эпителия и др.). Изменение структуры и числа хромосом (хромосомные аберрации) является признаком наследственной природы болезни.

Рис. 4.4. Схематическое изображение хромосом человека (идиограмма гаплоидного набора.

Рис. 4.5. Метафазная пластинка при простой окраске.
Молекулярно-генетический. Реализуется с помощью блот-гибритизации по Саузерну (введение флюоресцентной метки – ДНК-зонд) и амплификации (увеличении числа копий) участков ДНК при помощи ПЦР (полимеразной цепной реакции).
ПЦР осуществляется последовательными циклами. В каждом цикле происходят следующие события:
- двухспиральная ДНК при нагревании разделяется на составляющие одноцепочечные цепи и в таком состоянии может служить матрицей для репликации;
- далее одноцепочечные нити ДНК инкубируют в присутствии ДНК-полимеразы и раствора, содержащего смесь всех четырех нуклеотидов, а также специфические последовательности ДНК (праймеры), что приводит к синтезу копий двух молекул ДНК.
Затем процедуры повторяются сначала, и происходит копирование как старых, так и новых одноцепочечных цепей с образованием третьей и четвертой копий молекулы ДНК, затем все четыре снова копируются, и образуется уже восемь молекул ДНК, и т.д. число растет в геометрической прогрессии. В результате 20-30 циклов нарабатывается эффективное количество ДНК. Отдельный цикл занимает около 5 мин., а для бесклеточного молекулярного клонирования фрагмента ДНК требуется всего несколько часов.
Метод ПЦР отличается очень высокой чувствительностью: он позволяет обнаружить в пробе всего одну присутствующую в ней молекулу ДНК. Тот же способ пригоден и для анализа следовых последовательностей РНК, для этого РНК переводят в последовательности комплементарной ДНК (кДНК), используя обратную транскриптазу. Метод получил широкое использование в пренатальной диагностике наследственных болезней, выявлении вирусных инфекций, а также в судебной медицине.
VI. Мутагены. Классификация мутаций. Обозначения, принятые для изображения генеалогического дерева. Типы наследования дефектов в геноме.
Мутация – скачкообразное изменение признака вследствие количественных или качественных изменений генотипа.
Мутагены – факторы, вызывающие мутации.
Мутациибывают:
· соматические (потенциальное развитие опухолей) не передаются по наследству и, следовательно, не относятся к наследственным заболеваниям, хотя и поражают генетический аппарат клетки;
· гаметические (передаются по наследству).
§ Летальные – сопровождаются гибелью организма внутриутробно, или сразу после рождения.
§ Сублетальные – гибель до полового созревания.
§ Гипогенитальные – сочетаются с бесплодием.
По характеру изменения генотипав соответствии с тремя уровнями организации генетического материала (гены – хромосомы – геном) различают мутации – генные, хромосомные, геномные и цитоплазматические.
I. Генные мутации связаны с изменением структуры отдельных генов (участков ДНК, кодирующих синтез одного белка, одного признака).
· Моногенная – мутация в одном гене с изменением одного признака (например, альбинизм, короткопалость); моногенные мутации обуславливают истинные наследственные заболевания.
· Полигенные – одновременные мутации в различных генах различных хромосом, обуславливающие однонаправленные изменения в организме, которые определяют предрасположенность к некоторым заболеваниям (например, атеросклероз, гипертоническая болезнь, сахарный диабет II типа); наследуется не сама болезнь, а предрасположенность к ней, реализующаяся при воздействии определенных внешних факторов. Заболевание развивается как под влиянием мутаций, так и под влиянием факторов среды, т. е. является мультифакториальным. Даже для одного и того же заболевания относительное значение наследственности и среды у разных лиц может быть неодинаковым.
· Точковая – повреждение одного нуклеотида в гене, т. е. замена одной аминокислоты в белке на другую (например, ферментопатии, серповидно-клеточная анемия, глухонемота).
II. Хромосомные мутации- структурные перестройки в отдельных хромосомах: делеции, дупликации, инверсии, транслокации.
· Делеция - это потеря части хромосомы в результате ее разрыва. Большинство делеций летальны в результате потери огромной части генетического материала. Делеция короткого плеча 4 хромосомы приводит к развитию синдрома Вольфа; делеция короткого плеча 5 хромосомы - синдрома кошачьего крика (сri du chat)- мяуканье и звуки подобные кошачьему крику типичны для этой патологии, часто наблюдается отставание в умственном развитии и пороки сердца.
· Транслокация - это перенос отдельного сегмента одной хромосомы в другую хромосому. При сбаласнсированной транслокации весь генетический материал сохраняется и остается фунционально способным, поэтому фенотипических проявлений нет. У таких людей могут формироваться аномальные гаметы.
· Дупликация – удвоение участка хромосомы.
· Инверсии – поворот участка хромосомы на 1800.

Рис. 4.6. Схематическое изображение различных видов хромосомных мутаций.
III. Геномные мутации - изменения числа хромосом в наборе, не сопровождаемые изменением их структуры. Число хромосом при этом меняется некратно – формируется анеуплоидный набор хромосом. Кратное изменение числа хромосом (полиплоидия) несовместимо с жизнью.
1. Моносомии – уменьшение количества хромосом.
· Синдром Шерешевского-Тернера (яичниковая дисгенезия) - моносомия половых хромосом, встречается довольно часто. Отсутствует одна Х-хромосома (45, ХО). В некоторых случаях вторая Х-хромосома присутствует, но в ней выявляются тяжелые нарушения (изохромосома, частичная делеция и др.). Потеря второй Х-хромосомы обычно приводит к гибели плода.
У выживших детей наблюдается лимфэдема шеи, которая присутствует и у взрослых, приводя к формированию толстой шеи. Часто наблюдаются врожденные аномалии сердца, низкий рост, ожирение и нарушения строения скелета. Интеллект не нарушен. В присутствии одной Х-хромосомы (и отсутствии Y-хромосомы) примитивные половые железы развиваются как яичники. Отсутствие второй Х-хромосомы приводит к нарушению развития яичников в пубертатном периоде. Яичники остаются маленькими и в них не обнаруживаются примордиальные фолликулы. Также нарушается синтез эстрогенов, что проявляется нарушением эндометриального цикла (аменоррея) и слабым развитием женских вторичных половых признаков. Диагноз может быть поставлен при отсутствии телец Барра в соскобах буккального эпителия у лиц, имеющих женский фенотип или при анализе кариотипа.
· При аутосомной моносомии теряется огромное количество генетического материала, поэтому она обычно летальна.
2. Триосомии – увеличение количества хромосом на одну.
· Синдром Кляйнфельтера (тестикулярная дисгенезия) - трисомия половых хромосом - встречается довольно часто. Проявляется наличием лишней Х-хромосомы (47, ХХY), реже больные с синдромом Кляйнфельтера могут иметь две и более лишних Х-хромосом (48, ХХХY или 49, ХХХХY). Формируется мужской фенотип.
До пубертатного периода никаких клинических проявлений не наблюдается. Лишняя Х-хромосома нарушает нормальное развитие яичек в пубертатном периоде неизвестным способом. Яички остаются маленькими и не продуцируют сперматозоиды, больные обычно бесплодны. Уровень тестостерона в крови низкий, что приводит к нарушению развития вторичных половых признаков. У больных имеется склонность к высокому росту (тестостерон ускоряет окостенение эпифизов) и евнухоидный внешний вид с высоким голосом, маленьким пенисом и ростом волос по женскому типу. Также иногда наблюдается гинекомастия. Иногда наблюдается снижение интеллекта. Диагноз синдрома Кляйнфельтера может быть установлен при нахождении телец Барра в соскобах буккального эпителия у лиц, имеющих мужской фенотип или при анализе кариотипа.
· Синдром ХХХ (“суперженщины”)- присутствие третьей Х-хромосомы у женщин. Большинство пациентов являются нормальными. У некоторых наблюдаются нарушения умственного развития, нарушения менструального цикла и снижение фертильности (плодовитости).
· Синдром ХYY - присутствие лишней Y-хромосомы у мужчин. Большинство пациентов нормальные.
У некоторых может наблюдаться агрессивность поведения и легкое отставание в умственном развитии.
· Синдром Дауна является наиболее частым аутосомным нарушением. Он возникает в результате наличия третьей 21 хромосомы, что приводит к развитию характерных клинических проявлений.
Дети имеют характерный косой разрез глаз с уплощенным профилем, кососмотрящие глаза, резко выраженные вертикальные кожные складки, прикрывающие медиальный угол глазной щели, так называемое сходство с лицами азиатов, раньше называемое “монголоидным”. Постоянным признаком является отставание в умственном развитии. 30% пациентов имеют врожденные пороки сердца. Также у этих больных повышена заболеваемость различными инфекциями, язвами двенадцатиперстной кишки и острой лейкемией. Мужчины с синдромом Дауна обычно бесплодны, а женщины могут рожать детей. Потомки матерей с синдромом Дауна могут быть нормальными, потому что лишняя 21 хромосома содержится не во всех гаметах.

Рис. 4.7. Дети с синдромом Дауна.
· Синдром Эдвардса- трисомия 18 хромосомы (47ХХ/ХY, +18) встречается редко.
Клинически он проявляется отставанием в физическом и умственном развитии, сопровождаемым характерными физическими недостатками, такими как “стопа рокера” и сжатые в кулаки руки с перекрещивающимися пальцами. В результате тяжелых поражений дети редко выживают более года.
· Синдром Патау- трисомия 13 хромосомы (47ХХ/ХY, +13) также встречается редко. Большинство детей умирает сразу после рождения.
Трисомия 13 хромосомы характеризуется нарушением развития подкорковых структур мозга (отсутствие обонятельных луковиц, слияние лобных долей и единственный желудочек головного мозга) и срединных структур лица (расщепление губы, расщепление твердого неба, дефекты носа, единственный глаз [циклоп]).
IV. Цитоплазматические (митохондриальные) мутациивозникают в результате мутаций в плазмогенах, находящихся в ДНК-содержащих клеточных органеллах - митохондриях. Некоторые патологии, связанные приводящие к мужскому бесплодию, связаны с этим видом мутаций. Некоторые виды близнецовости могут быть обусловлены этими же причинами, при этом наследуются, как правило, только по женской линии.
В стандартной родословной используются простые условные обозначения и правила:
- Мужчины всегда изображаются в виде квадратов, женщины - в виде окружностей.
- Пациент, обратившийся к генетику для составления родословной - пробанд – обозначается стрелкой.
- Графически изображаемые связи между членами родословной бывают только трех видов: "мужья-жены", "дети-родители" и "братья-сестры".
- Супруги, братья и сестры (в т. ч. двоюродные и троюродные) всегда изображаются на одном горизонтальном уровне (т. е. в одном поколении). Разница в возрасте не играет никакой роли.
- Дети пробанда изображаются на горизонтальном уровне ниже пробанда, а его родители - на горизонтальном уровне выше пробанда. То же самое относится к детям и родителям всех братьев и сестер пробанда.
- Все поколения нумеруются сверху вниз римскими цифрами, а все индивидуумы в каждом поколении - слева направо арабскими цифрами. Это позволяет обозначить каждого человека личным идентификационным номером (например - III:15, что означает 15-й индивидуум в третьем поколении).

Рис. 4.8. Символы, используемые при составлении родословной.